
Abstract
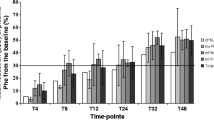
Treatment with tetrahydrobiopterin (BH4), the natural cofactor of phenylalanine hydroxylase (PAH), can reduce blood phenylalanine (Phe) levels in patients with BH4-responsive phenylketonuria (PKU). A number of studies has reported on the short-term BH4 treatment of patients with PKU, but long-term data are lacking. Here, we describe the effects of long-term treatment with BH4 on 16 patients, who showed a >28% reduction in blood Phe following testing for BH4 overload. The mean dose of BH4 was 16 mg/kg body weight (range 5–36 mg/kg body weight). The mean treatment duration was 56 months (range 24–110 months). Of 16 patients, 14 achieved long-term Phe control with BH4 treatment, with a mean blood Phe concentration of 321 ± 236 µmol/l. The mean decrease from baseline in blood Phe levels in these 14 patients was 54.6%. Of the seven patients who required continued dietary restriction, Phe intake increased from 200–300 mg/day to 800–1000 mg/day. Factors that may cause fluctuation of Phe levels in BH4-treated patients include patients’ PAH genotype, Phe intake, changes in protein catabolism or anabolism, and periods of illness or infection.



Similar content being viewed by others

Abbreviations
- BH4 :
-
tetrahydrobiopterin
- EC:
-
Enzyme Commission
- HAWIK:
-
Hamburg Wechsler Intelligenz-Test für Kinder
- MHPA:
-
mild hyperphenylalaninaemia
- OMIM:
-
Online Mendelian Inheritance in Man database
- PAH:
-
phenylalanine hydroxylase
- Phe:
-
phenylalanine
- PKU:
-
phenylketonuria
- RAMEDIS:
-
rare metabolic disease database www.ramedis.de
- SD:
-
standard deviation
- SEE:
-
standard error of estimation
References
Bélanger-Quintana A, García MJ, Castro M et al (2005) Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Genet Metab 86(Suppl 1):S61–S66
Burgard P, Rupp A, Konecki DS, Trefz FK, Schmidt H, Lichter-Konecki U (1996) Phenylalanine hydroxylase genotypes, predicted residual enzyme activity and phenotypic parameters of diagnosis and treatment of phenylketonuria. Eur J Pediatr 155(Suppl 1):S11–S15
Burgard P, Bremer HJ, Bührdel P et al (1999) Rationale for the German recommendations for phenylalanine level control in phenylketonuria 1997. Eur J Pediatr 158:46–54
Kure S, Hou DC, Ohura T et al (1999) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135:375–378
Lambruschini N, Pérez-Dueñas B, Vilaseca MA et al (2005) Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab 86(Suppl 1):S54–S60
Lee P, Treacy EP, Crombez E et al (2008) Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet 146A:2851–2859
Levy HL, Milanowski A, Chakrapani A et al (2007) Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 370:504–510
National Institutes of Health Consensus Development Panel (2001) National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics 108:972–982
Trefz FK, Scheible D, Frauendienst-Egger G, Korall H, Blau N (2005) Long-term treatment of patients with mild and classical phenylketonuria by tetrahydrobiopterin. Mol Genet Metab 86(Suppl 1):S75–S80
Trefz FK, Burton BK Longo N, Casanova MM et al (2009) Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 154:700–707
Zurflüh MR, Zschocke J, Lindner M et al (2008) Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat 29:167–175
Acknowledgements
The authors take full responsibility for the content of this article but thank Caudex Medical (supported by Serono Symposia International Foundation) for their assistance in preparing the initial draft of this report and collating the comments of the authors and any other named contributors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Nenad Blau
Reference to electronic databases: EC, OMIM, RAMEDIS
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Trefz, F.K., Scheible, D. & Frauendienst-Egger, G. Long-term follow-up of patients with phenylketonuria receiving tetrahydrobiopterin treatment. J Inherit Metab Dis 33 (Suppl 3), 163–169 (2010). https://doi.org/10.1007/s10545-010-9058-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9058-x