
Abstract
Objective
To perform the ultrastructural examination of a chorionic villi biopsy as a predictor of foetal involvement in the infantile form of glycogenosis type II (Pompe disease).
Methods
Ultrastructural, biochemical and genetic analyses were performed on chorionic villi biopsies of three consecutive pregnancies in a woman with a previous child affected by Pompe disease.
Results
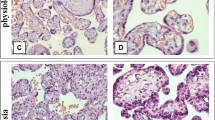
In the only affected foetus, glycogen storage was observed in fibrocytes and endothelial cells of a chorionic villi sample at 11 week’s gestation. Severe multi-organ involvement was demonstrated in the tissues of the aborted foetus. No abnormal material was found in the chorionic samples of two subsequent pregnancies, and a healthy boy and girl were born at term and remain unaffected. Both exhibited a partial reduction in acid maltase and were carriers of the maternal mutation.
Conclusions
Ultrastructural findings correlated with biochemical and genetic results, providing a clear and early indicator of the definite diagnosis for future pregnancy management or an early therapeutic approach.


Similar content being viewed by others


Abbreviations
- GSDII:
-
glycogen storage disease type II
- ERT:
-
enzyme replacement therapy
- CK:
-
creatine kinase
- EMG:
-
electromyography
- PAS:
-
periodic acid–Schiff
- GAA :
-
acid α-glucosidase gene
- 4MUG:
-
4-methylumbelliferyl-α-D-glucoside
- CVS:
-
chorionic villi sample
- LSDs:
-
lysosomal storage disorders
References
Bembi B, Cerini E, Danesino C, Donati MA, Gasperini S, Morandi L, Musumeci O, Parenti G, Ravaglia S, Seidita F, Toscano A, Vianello A (2008) Diagnosis of glycogenosis type II. Neurology 71:S4–S11
Bendon RW, Hug G (1985) Morphologic characteristics of the placenta in glycogen storage disease type II (alpha-1,4-glucosidase deficiency). Am J Obstet Gynecol 152:1021–1026
Fernandez-Hojas R, Huie ML, Navarro C et al (2002) Identification of six novel mutations in the acid alpha-glucosidase gene in three Spanish patients with infantile onset glycogen storage disease type II (Pompe disease). Neuromuscul Disord 12:159–166
Fowler DJ, Anderson G, Vellodi A, Malone M, Sebire NJ (2007) Electron microscopy of chorionic villus samples for prenatal diagnosis of lysosomal storage disorders. Ultrastruct Pathol 31:15–21
Hesselink RP, Schaart G, Wagenmakers AJ, Drost MR, van der Vusse GJ (2006) Age-related morphological changes in skeletal muscle cells of acid alpha-glucosidase knockout mice. Muscle Nerve 233:505–513
Hirschhorn R, Reuser AJJ (2001) Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Valle D, Sly WS (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 3389–3420
Hug G, Chuck G, Chen YT, Kay HH, Bossen EH (1991) Chorionic villus ultrastructure in type II glycogen storage disease (Pompe’s disease). N Engl J Med 324:342–343
Konstantinidou AE, Anninos H, Dertinger S, Nonni A, Petersen M, Karadimas C, Havaki S, Marinos E, Akman HO, DiMauro S, Patsouris E (2008) Placental involvement in glycogen storage disease type IV. Placenta 29(4):378–381
Infantile-Onset Pompe Disease Natural History Study Group, Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D (2006) A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 148:671–676
Litwin JA, Kasprzyk JM (1976) PAS reaction performed on semithin epon sections following removal of the resin by NaOH in absolute ethanol. Acta Histochem 55:98–103
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Martin JJ, Ceuterick C, Leroy JG (1976) Skin biopsy in the diagnosis of metabolic encephalopathies. Rev Neurol (Paris) 132:639–651
McCready ME, Carson NL, Chakraborty P, Clarke JT, Callahan JW, Skomorowski MA, Chan AK, Bamforth F, Casey R, Rupar CA, Geraghty MT (2007) Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II. Mol Genet Metab 92:325–335
O’Brien JS, Bernett J, Veath ML, Paa D (1975) Lysosomal storage disorders. Diagnosis by ultrastructural examination of skin biopsy specimens. Arch Neurol 32:592–599
Park HK, Kay HH, McConkie-Rosell A, Lanman J, Chen YT (1992) Prenatal diagnosis of Pompe’s disease (type II glycogenosis) in chorionic villus biopsy using maltose as a substrate. Prenat Diagn 12:169–173
Phupong V, Shuangshoti S, Sutthiruangwong P, Maneesri S, Nuayboonma P, Shotelersuk V (2005) Prenatal diagnosis of Pompe disease by electron microscopy. Arch Gynecol Obstet 271:259–261
Raben N, Fukuda T, Gilbert AL et al (2005) Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol Ther 11:48–56
Shanske S, DiMauro S (1981) Late-onset acid maltase deficiency. Biochemical studies in leukocytes. J Neurol Sci 50:57–62
van den Hout HM, Hop W, van Diggelen OP et al (2003) The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 112:332–340
Wierzba-Bobrowicz T, Lewandowska E, Lugowska A et al (2007) Adult glycogenosis type II (Pompe’s disease): morphological abnormalities in muscle and skin biopsies compared with acid alpha-glucosidase activity. Folia Neuropathol 45:179–186
Acknowledgements
The authors are grateful to Christine O’Hara for her help with the English version of the paper, to Soraya Barrera for her excellent technical work, and to Tania Vazquez for editorial assistance. This work was supported by grants from Fondo de Investigacion Sanitaria (PI07/1257 and PI07/90043) and Xunta de Galicia (PGIDIT06PXIB905328PR and INCITE07PXI905221ES). Beatriz San Millan was supported by a research contract from the Instituto de Salud Carlos III (ISCIII), (Exp CM07/00135).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Alberto B. Burlina
Competing interest: None declared.
Beatriz San Millan and Susana Teijeira contributed in equal measure to this study
References to electronic databases: OMIM catalogue number: Pompe disease, glycogen storage disease type II, OMIM 232300.
Rights and permissions
About this article
Cite this article
San Millan, B., Teijeira, S., Domínguez, C. et al. Chorionic villi ultrastructure in the prenatal diagnosis of glycogenosis type II. J Inherit Metab Dis 33 (Suppl 3), 105–111 (2010). https://doi.org/10.1007/s10545-009-9033-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-009-9033-6