
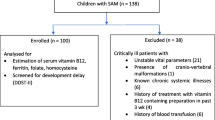
Abstract
A pilot expanded newborn screening programme to detect inherited metabolic disorders by means of liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) began in the Campania region, southern Italy, in 2007. By October 2009, >8,800 dried blood samples on filter paper from 11 hospitals had been screened. Within this screening programme, we identified a case of mitochondrial acetoacetyl-coenzyme A (CoA) thiolase deficiency [β-ketothiolase (β-KT) deficiency] by analysing the acylcarnitine profile from a dried blood spot with LC-MS/MS. Gas chromatography coupled with mass spectrometry analysis of urinary organic acids and LC-MS/MS analysis of urinary acylcarnitines were in line with this disorder. In fact, concentrations were well beyond the cut-off values of tiglyl carnitine, 3-hydroxybutyrylcarnitine and 2-methyl-3-hydroxybutyrylcarnitine, 2-methyl-3-hydroxybutyric acid and tiglyl glycine. The absence of 2-methylacetoacetic acid in urine may be attributed to: (i) the instability of this β-ketoacid because it undergoes spontaneous decarboxylation to 2-butanone, which is highly volatile and thus difficult to detect, and (ii) the good health of the patient in the first days of life. β-KT deficiency was subsequently diagnosed in the patient's older sister, who showed increased levels of the same metabolites but also small amounts of 2-methylacetoacetic acid, which is considered a key marker for β-KT diagnosis. Genomic analysis revealed mutation c.1189C >G in exon 12 of the ACAT1 gene, which results in a severe defect because of the p.H397D amino acid change in both alleles of both patients.
Similar content being viewed by others


Introduction
A pilot expanded newborn screening programme to detect inherited metabolic disorders by means of liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) began in the Campania region, southern Italy, in 2007. By October 2009, >8,800 dried blood samples on filter paper from 11 hospitals in the region had been screened in our laboratory. Within this programme, we detected mitochondrial acetoacetyl-coenzyme A (CoA) thiolase deficiency (β-KT deficiency) in a newborn asymptomatic girl and in her older sister who had a metabolic decompensation event a few months earlier (Fig. 1). In the proband, β-KT deficiency was diagnosed by analysing the patient’s acylcarnitine profile on a dried blood spot and in urine by using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) and gas chromatography coupled with mass spectrometry (GC/MS) using previously described procedures (Chace 2009; Millington et al. 1991; Mueller et al. 2003).

Dual function of acetoacetyl-coenzyme A (CoA) thiolase [ACAT 1 (β-KT)] in isoleucine and ketone body metabolism. Metabolite biomarkers of β-KT deficiency are boxed
Results
Patient 1
The proband, a girl, was born after 41 weeks of an uncomplicated pregnancy, the second child of healthy parents who were third cousins. Birth weight was 3,390 g (50th centile), length 53 cm (50th–75th centile) and head circumference 36 cm (50th–75th centile). No clinical abnormalities were found in the first days of life. The Apgar score was 9 at 1 min and 10 at 5 min. The mother was 25 years old and the father 27. The amino acid profile determined in a dried blood spot when patient 1 was 4 days old was normal, whereas acylcarnitine analysis revealed increased concentrations of tiglylcarnitine (C5:1 = 0.34 μmol/L; upper cut-off = 0.23 μmol/L), 3-hydroxybutyrylcarnitine (C4OH = 2.19 μmol/L; upper cut-off = 0.55 μmol/L) and 2-methyl-3-hydroxybutyrylcarnitine (C5OH = 1.17 μmol/L; upper cut-off = 0.28 μmol/L). Analysis of acylcarnitines in urine samples confirmed elevated concentrations of C5:1 (2.68 mmol/mol creatinine; upper cut-off = 0.20 mmol/mol creatinine) and C5OH (1.08 mmol/mol creatinine; upper cut-off = 0.54 mmol/mol creatinine). C5:1 and C5OH concentrations are elevated in both β-KT deficiency and 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (MHBD deficiency) (Matern et al 2003), whereas increased concentrations of C4OH have been identified only in cases of β-KT deficiency (Han et al 2007 and http://region4genetics.org). Therefore, elevated C4OH concentrations were highly suggestive of β-KT deficiency in patient 1. To verify the diagnosis, we analysed urine organic acids. Results showed that patient 1 had abnormal excretion of 2-methyl-3-hydroxybutyric acid (441 mmol/mol creatinine; upper cut-off = 2 mmol/mol creatinine) and tiglylglycine (87 mmol/mol creatinine; upper cut-off = 2 mmol/mol creatinine). Detection of 2-methylacetoacetic acid in the urine organic acid profile is considered a key factor for a diagnosis of β-KT deficiency; 2-methylacetoacetic acid is unstable because it undergoes spontaneous decarboxylation to 2-butanone, which is highly volatile and is therefore not easily determined (Korman 2006). Its absence in the urine of patient 1 can probably be attributed to her good health because of the earlier diagnosis consequent to neonatal screening.
Patient 2
Birth weight of the older sister of patient 1 was 3,240 g (25th centile). She was born after an uncomplicated pregnancy. At 18 months of age, after gastroenteritis and a vomiting event, she entered an emergency unit because of lethargy and metabolic acidosis (pH 7.07, HCO3 - 5.2). The metabolic decompensation was treated with bicarbonate infusion. β-KT deficiency was diagnosed when the patient was 23 months old and in an asymptomatic condition soon after the metabolic defect was discovered in her sister. Serum and urine acylcarnitine analysis in patient 2 revealed elevated concentrations of both C5:1 (serum: 0.96 μmol/L with an upper cut-off = 0.12 μmol/L; urine: 12.8 mmol/mol creatinine with an upper cut-off = 0.2 mmol/mol creatinine) and C5OH (serum: 0.70 μmol/L with an upper cut-off = 0.13 μmol/L; urine: 2.06 mmol/mol creatinine with an upper cut-off = 0.54 mmol/mol creatinine). Urine organic acid analysis revealed massive excretion of 2-methyl-3-hydroxybutyric acid (1,354 mmol/mol creatinine; upper cut-off = 2 mmol/mol creatinine) and tiglylglycine (825 mmol/mol creatinine; upper cut-off = 2 mmol/mol creatinine) and small amounts of 2-methylacetoacetic acid (25 mmol/mol creatinine). Thus, taken together, the data suggested β-KT deficiency in this patient and indirectly confirmed the same diagnosis in the younger sister.
Familial genomic analysis
To reach a more robust diagnosis, we carried out a genomic analysis in both sisters and their parents. We sequenced the amplified fragments corresponding to exons 1–12 of the ACAT1 gene and identified the c.471C >A p.S547S (rs35188041) polymorphism in exon 6 and the c.1189C >G p.H397D mutation in exon 12, both at the homozygous state in the two patients. The parents were found to be heterozygous for both the polymorphism and the mutation, thus confirming the homozygosity of the two daughters. The H397D mutation changes an amino acid residue that is highly conserved among species. The H397D mutation provokes a null allele in the sense that it causes the total loss of acetoacetyl-CoA thiolase activity for which the potassium ion is critical (Fukao et al 2003, 2008; Haapalainen et al 2007). Such a severe three-dimensional perturbation destabilises the overall integrity of the mutant enzyme (Zhang et al 2004).
Discussion
Here we report the clinical and biological investigations carried out in two sisters in whom we identified β-KT deficiency. Using LC-MS/MS within a pilot extended metabolic screening programme, we diagnosed β-KT deficiency by analysing the blood spot of patient 1. Subsequently, we identified the same metabolic defect in the patient’s sister by analysing a serum sample. In the next level of diagnosis, we examined urinary organic acids using GC/MS in both sisters. This analysis revealed abnormal excretion of 2-methyl-3-hydroxybutyric acid and tiglylglycine in both patients and a small amount of 2-methylacetoacetic acid in the older sister. Familial genomic analysis confirmed that both sisters are affected by β-KT deficiency and showed that they are homozygotes for the c.1189C >G (p.H397D) mutation in exon 12 of the ACAT1 gene; the parents are heterozygotes for the same mutation. The mutation has been classified as a severe defect because it abolishes protein expression, as demonstrated in an in vitro assay (Zhang et al 2004).
Abbreviations
- β-KT:
-
β-ketothiolase
- C5:1:
-
Tiglylcarnitine
- C4OH:
-
3-hydroxybutyrylcarnitine
- C5OH:
-
2-methyl-3-hydroxybutyrylcarnitine
- GC/MS:
-
Gas chromatography coupled with mass spectrometry
- LC-MS/MS:
-
Liquid chromatography coupled with tandem mass spectrometry
- MHBD:
-
2-methyl-3-hydroxybutyrylCoA dehydrogenase deficiency
References
Chace D (2009) Mass spectrometry in newborn and metabolic screening: historical perspective and future directions. J Mass Spectrometry 44:163–170
Fukao T, Zhang GX, Sakura N et al (2003) The mitochondrial acetoacetyl-CoA thiolase (T2) deficiency in Japanese patients: urinary organic acid and blood acylcarnitine profiles under stable conditions have subtle abnormalities in T2-deficient patients with some residual T2 activity. J Inherit Metab Dis 26:423–431
Fukao T, Boneh A, Aoki Y, Kondo N (2008) A novel single-base substitution (c.1124A >G) that activates a 5-base upstream cryptic splice donor site within exon 11 in the human mitochondrial acetoacetyl-CoA thiolase gene. Molec Genet Metab 94:417–421
Haapalainen A, Merilainen G, Pirila P, Kondo N, Fukao T, Wierenga R (2007) Crystallographic and kinetic studies of human mitochondrial acetoacetyl-CoA thiolase: the importance of potassium and chloride ions for its structure and function. Biochemistry 46:4305–4321
Han LS, Ye J, Qiu WJ, Gao XL, Wang Y, Gu XF (2007) Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis 30:507–514
Korman SH (2006) Inborn errors of isoleucine degradation: a review. Molec Genet Metab 89:289–99
Matern D, He M, Berry S et al (2003) Prospective diagnosis of 2-Methylbutyryl-CoA dehydrogenase deficiency in the Hmong population by newborn screening using tandem mass spectrometry. Pediatrics 112:74–78
Millington SD, et al (1991) The analysis of diagnostic markers of genetic disorders in human blood and urine using tandem mass spectrometry with liquid secondary ion mass spectrometry. Int J Mass Spectrom Ion Process 111:211–228
Mueller P, Schulze A, Schindler I, Ethofer T, Buehrdel P, Ceglarek U (2003) Validation of an ESI-MS/MS screening method for acylcarnitine profiling in urine specimens of neonates, children, adolescents and adults. Clin Chim Acta 327:47–57
Zhang GX, Fukao T, Mo R et al (2004) Mitochodrial acetoacetyl-CoA Thiolase (T2) deficiency: T2-deficient patients with “mild” mutation(s) were previously misinterpreted as normal by the coupled assay with tiglyl-CoA. Pediatr Res 56:60–64
Acknowledgements
This work was supported by grants from Regione Campania (DGRC 2362/07). We thank Jean Ann Gilder for editing the text.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: K. Michael Gibson
Catanzano and Ombrone contributed equally to the study.
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Catanzano, F., Ombrone, D., Di Stefano, C. et al. The first case of mitochondrial acetoacetyl-CoA thiolase deficiency identified by expanded newborn metabolic screening in Italy: the importance of an integrated diagnostic approach. J Inherit Metab Dis 33 (Suppl 3), 91–94 (2010). https://doi.org/10.1007/s10545-009-9028-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-009-9028-3