
Summary
Background:
The clinical severity of phenylalanine hydroxylase deficiency is usually defined by either pre-treatment phenylalanine (Phe) concentration or Phe tolerance at 5 years of age. So far, little is known about the course of Phe tolerance or the ability of both pre-treatment Phe and Phe tolerance at early age to predict Phe tolerance at later age.
Aim:
This study was conducted to investigate the course of the individual Phe tolerance and to assess the predictive value of both the pre-treatment Phe concentration and Phe tolerance at 1 and 6 months and 1, 2, 3 and 5 years for Phe tolerance at 10 years of age.
Method:
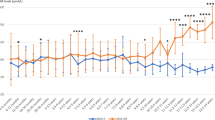
Data on blood Phe concentration, prescribed Phe intake and weight of 213 early and continuously treated Dutch PKU patients up to 10 years of age were collected. Data acquired under good metabolic control were used in the study. Tolerance was expressed in mg/day and mg/kg per day.
Results:
Data at 1 and 6 months and at 1, 2, 3 and 5 years of 61, 58, 59, 57, 56 and 59 patients were included for comparison with the Phe tolerance at 10 years. Phe tolerances (mg/kg per day) at 2, 3 and 5 years showed a clear correlation with the tolerance at 10 years of age (r = 0.608, r = 0.725 and r = 0.661). Results for tolerance expressed as mg/day were comparable. Pre-treatment Phe concentrations did not correlate significantly with the tolerance.
Conclusion:
Pre-treatment Phe is unreliable but Phe tolerance is a reliable predictor of the tolerance at 10 years of age, starting at 2 years of age.
Similar content being viewed by others


Abbreviations
- BH4 :
-
tetrahydrobiopterin
- PAH:
-
phenylalanine hydroxylase
- PAL:
-
phenylalanine ammonia-lyase
- Phe:
-
phenylalanine
- PKU:
-
phenylketonuria
References
Burgard P, Rupp A, Konecki DS, Trefz FK, Schmidt H, Lichter-Konecki U (1996) Phenylalanine hydroxylase genotypes, predicted residual enzyme activity and phenotypic parameters of diagnosis and treatment of phenylketonuria. Eur J Pediatr 155(Supplement 1): S11–S15. doi:10.1007/PL00014222.
Burton BK, Grange DK, Milanowski A, et al (2007) The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 30: 700–707. doi:10.1007/s10545-007-0605-z.
Crone MR, van Spronsen FJ, Oudshoorn K, Bekhof J, van Rijn G, Verkerk PH (2005) Behavioural factors related to metabolic control in patients with phenylketonuria. J Inherit Metab Dis 28: 627–637. doi:10.1007/s10545-005-0014-0.
Goris AH, Westerterp KR (2000) Improved reporting of habitual food intake after confrontation with earlier results on food reporting. Br J Nutr 83: 363–369.
Guldberg P, Rey F, Zschocke J, et al (1998) A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet 63: 71–79. doi:10.1086/301920.
Güttler F (1980) Hyperphenylalaninemia: diagnosis and classification of the various types of phenylalanine hydroxylase deficiency in childhood. Acta Paediatr Scand Suppl 280: 1–80.
Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR (1997) Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype–phenotype correlations. Am J Hum Genet 61: 1309–1317. doi:10.1086/301638.
Levy HL, Milanowski A, Chakrapani A, et al (2007) Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 370: 504–510. doi:10.1016/S0140-6736(07)61234-3.
Lichter-Konecki U, Rupp A, Konecki DS, Trefz FK, Schmidt H, Burgard P (1994) Relation between phenylalanine hydroxylase genotypes and phenotypic parameters of diagnosis and treatment of hyperphenylalaninaemic disorders. German Collaborative Study of PKU. J Inherit Metab Dis 17: 362–365. doi:10.1007/BF00711831.
MacDonald A, Rylance G, Hall SK, Asplin D, Booth IW (1996) Factors affecting the variation in plasma phenylalanine in patients with phenylketonuria on diet. Arch Dis Child 74: 412–417.
Medical Research Council (1993) Recommendations on the dietary management of phenylketonuria. Report of Medical Research Council Working Party on Phenylketonuria. Arch Dis Child 68: 426–427.
Rey F, Blandin-Savoja F, Rey J (1979) Kinetics of phenylalanine disappearance after intravenous load in phenylketonuria and its genetic variants. Pediatr Res 13: 21–25. doi:10.1203/00006450-197901000-00005.
Sarkissian CN, Gamez A (2005) Phenylalanine ammonia lyase, enzyme substitution therapy for phenylketonuria, where are we now? Mol Genet Metab 86(Supplement 1): S22–S26. doi:10.1016/j.ymgme.2005.06.016.
Stemerdink BA, Kalverboer AF, van der Meere JJ, et al (2000) Behaviour and school achievement in patients with early and continuously treated phenylketonuria. J Inherit Metab Dis 23: 548–562. doi:10.1023/A:1005669610722.
Treacy EP, Delente JJ, Elkas G, et al (1997) Analysis of phenylalanine hydroxylase genotypes and hyperphenylalaninemia phenotypes using l-[1-13C]phenylalanine oxidation rates in vivo: a pilot study. Pediatr Res 42: 430–435. doi:10.1203/00006450-199710000-00002.
Trefz FK, Batzler U, Konig T, et al (1990) Significance of the in vivo deuterated phenylalanine load for long-term phenylalanine tolerance and psycho-intellectual outcome in patients with PKU. Eur J Pediatr 149(Supplement 1): S25–S27. doi:10.1007/BF02126295.
Verkerk PH, Vaandrager GJ, Sengers RC (1990) 15 years of national screening for phenylketonuria in The Netherlands; 4th Report of the National Commission for Management of Phenylketonuria. Ned Tijdschr Geneeskd 134: 2533–2536.
Walter JH, White FJ (2004) Blood phenylalanine control in adolescents with phenylketonuria. Int J Adolesc Med Health 16: 41–45.
Acknowledgements
The authors express their gratitude to the dieticians of all metabolic departments of the university clinics who supplied dietary data on the PKU patients.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Nenad Blau
Competing interests: None declared
References to electronic databases: Phenylalanine hydroxylase: EC 1.14.16.1.
Rights and permissions
About this article
Cite this article
van Spronsen, F.J., van Rijn, M., Dorgelo, B. et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J Inherit Metab Dis 32, 27–31 (2009). https://doi.org/10.1007/s10545-008-0937-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-008-0937-3