
Summary
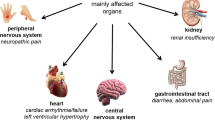
Background: Fabry disease (OMIM 301500) is an X-linked lysosomal storage disorder with characteristic vascular, renal, cardiac and cerebral complications. Globotriaosylceramide (Gb3) accumulates in Fabry patients as a result of α-galactosidase A deficiency. The phenotypic variability is high, but the relationship between clinical symptoms in individual Fabry patients has not been uniformly documented. Also, the relation between the most prominent biochemical abnormalities, elevated Gb3 levels in plasma and urine, and clinical symptoms is not firmly established. Methods: Clinical and biochemical characteristics of 96 (25 deceased) Dutch Fabry patients were collected retrospectively and before the initiation of enzyme therapy. Results: Clinical assessment revealed that median life expectancy was 57 years for male and 72 years for female patients. Cerebral complications, acroparaesthesias and gastrointestinal complications, but not cardiac and auditory complications, were all seen more frequently in male than female patients. Glomerular filtration rate (GFR) was highly variable in male patients, including 2 patients with GFR < 30 ml/min, but median GFR did not differ between males and females (103 and 101 ml/min, respectively). Hyperfiltration was more frequently observed in the female patient group. Microalbuminuria was present in 60% of males and 45% of females. No specific pattern of combined symptoms existed except for a relationship between left ventricular hypertrophy (LVH) and cerebral complications (males 36%, females 32%), or proteinuria (males 35%, females 31%). Gb3 was found to be more elevated in plasma samples from male (n = 26; median 6.27 μmol/L (1.39–9.74)) than female Fabry patients (n = 37; median 2.16 (0.77–4.18)). This was also observed for urinary Gb3: males (n = 22) median 1851 nmol/24 h (40–3724); females (n = 29) median 672 (86–2052). Plasma and urinary Gb3 levels correlated with each other in both males (r = 0.4, p = 0.05) and females (r = 0.4, p = 0.03), but no correlation between elevated Gb3 levels and clinical symptoms could be detected. Conclusion: Analysis of the characteristics of the Dutch Fabry cohort has revealed that a limited relationship between various disease manifestations exists and that individual symptoms do not correlate with elevated urinary or plasma Gb3 levels, limiting their value as surrogate disease markers.
Similar content being viewed by others


Abbreviations
- αGal A:
-
α-galactosidase A
- BPI:
-
Brief Pain Inventory
- CVA:
-
cerebrovascular accident
- ECG:
-
electrocardiogram
- ERPF:
-
effective renal plasma flow
- FF:
-
filtration fraction
- Gb3 :
-
globotriaosylceramide
- GFR:
-
glomerular filtration rate
- HPLC:
-
high-pressure liquid chromatography
- K/DOQI:
-
kidney disease outcomes quality initiative
- LVH:
-
left ventricular hypertrophy
- LVMass:
-
left ventricular mass
- MRI:
-
magnetic resonance imaging
- MSSI:
-
Mainz severity score index
- PTA:
-
pure-tone average
References
Apperloo AJ, de Zeeuw D, Donker AJ, de Jong PE (1996) Precision of glomerular filtration rate determinations for long-term slope calculations is improved by simultaneous infusion of 125I-iothalamate and 131I-hippuran. J Am Soc Nephrol 7: 567–572.
Bikkina M, Levy D, Evans JC, et al (1994) Left ventricular mass and risk of stroke in an elderly cohort. The Framingham Heart Study. JAMA 272: 33–36.
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med 276: 1163–1167.
Branton MH, Schiffmann R, Sabnis SG, et al (2002) Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81: 122–138.
de Groot WP (1961) Thesaurismosis heriditaria Ruiter–Pompen–Wyers: (Angiokeratoma corporis diffusum Fabry). University of Amsterdam, Amsterdam.
de Zeeuw D, Hillege HL, de Jong PE (2005) The kidney, a cardiovascular risk marker, and a new target for therapy. Kidney Int Suppl 98:S25–S29.
Deegan P, Baehner AF, Barba-Romero MA, et al (2006) Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 43(4): 347–352.
Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W (1973) Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med 81: 157–171.
Desnick RJ, Ioannou YA, Eng ME (2001) a-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds; Childs B, Kinzler KW, Vogelstein B, assoc, eds. The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 733–3774.
Eng CM, Guffon N, Wilcox WR, et al (2001) Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med 345: 9–16.
Folch J, Lees M, Sloane-Stanley GH (1957) A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226: 497–509.
Gerstein HC, Mann JF, Yi Q, et al (2001) Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 286: 421–426.
Guffon N (2003) Clinical presentation in female patients with Fabry disease. J Med Genet 40: e38.
Gupta S, Ries M, Kotsopoulos S, Schiffmann R (2005) The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore) 84: 261–268.
K/DOQI (2002) Clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 39: S1–266.
Kint JA (1970) Fabry’s disease: alpha-galactosidase deficiency. Science 167: 1268–1269.
Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP (1987) Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol 59: 956–960.
Linthorst GE, Hollak CEM (2005) Angiokeratomas, Fabry disease and enzyme replacement therapy: still a challenge: reply from authors. Br J Dermatol 152: 178–179.
MacDermot KD, Holmes A, Miners AH (2001a) Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 38: 769–775.
MacDermot KD, Holmes A, Miners AH (2001b) Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet 38: 750–760.
Mayes JS, Scheerer JB, Sifers RN, Donaldson ML (1981) Differential assay for lysosomal α-galactosidases in human tissues and its application to Fabry’s disease. Clin Chim Acta 112: 247–251.
Mehta A, Ricci R, Widmer U, et al (2004) Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 34: 236–242.
Merrill AH, Jr., Wang E, Mullins RE, Jamison WC, Nimkar S, Liotta DC (1988) Quantitation of free sphingosine in liver by high-performance liquid chromatography. Anal Biochem 171: 373–381.
NIH Technology Assessment Panel on Gaucher Disease (1996) Gaucher disease. Current issues in diagnosis and treatment. JAMA 275: 548–553.
Redonnet-Vernhet I, Ploos van Amstel JK, Jansen RP, Wevers RA, Salvayre R, Levade T (1996) Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene. J Med Genet 33: 682–688.
Schiffmann R, Kopp JB, Austin HA, III, et al (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285: 2743–2749.
Selvetella G, Notte A, Maffei A, et al (2003) Left ventricular hypertrophy is associated with asymptomatic cerebral damage in hypertensive patients. Stroke 34: 1766–1770.
Taketomi T, Hara A, Uemura K, Sugiyama E (1996) Rapid method of preparation of lysoglycosphingolipids and their confirmation by delayed extraction matrix-assisted laser desorption ionization time-of-flight mass spectrometry. J Biochem (Tokyo) 120: 573–579.
Vedder AC, Strijland A, vd Bergh Weerman MA, Florquin S, Aerts JM, Hollak CE (2006) Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis 29: 106–111.
Ware J, Snow K, Kosinski M, Gandek B (1997) SF36 Health Survey: manual and interpretation guide. Health Institute, New England Medical Center, Boston.
Whybra C, Kampmann C, Willers I, et al (2001) Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis 24: 715–724.
Whybra C, Kampmann C, Krummenauer F, et al (2004) The Mainz Severity Score Index: a new instrument for quantifying the Anderson–Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet 65: 299–307.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Joe Clarke
Competing interests: None declared
Rights and permissions
About this article
Cite this article
Vedder, A.C., Linthorst, G.E., van Breemen, M.J. et al. The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis 30, 68–78 (2007). https://doi.org/10.1007/s10545-006-0484-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-006-0484-8