
Summary
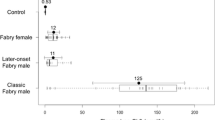
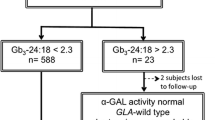
Anderson-Fabry disease (referred to as Fabry disease) is an X-linked disorder characterized by a deficiency of the lysosomal enzyme α-galactosidase A and the subsequent accumulation in various tissues of globotriaosylceramide (Gb3), the main substrate of the defective enzyme. Enzyme replacement therapy (ERT) offers a specific treatment for patients with Fabry disease, though monitoring of treatment is hampered by a lack of surrogate markers of response. In this study, the efficacy of long-term ERT in six Fabry hemizygotes and two symptomatic heterozygotes has been evaluated. Patients were administered recombinant α-galactosidase A every 2 weeks for up to a year. The efficacy of ERT was assessed by monitoring symptomatology and renal function. Urinary glycolipid concentration was estimated by a novel tandem mass spectrometric method. Urine glycolipid (Gb3) was elevated at baseline and fell impressively on ERT where patients were hemizygotes and in the absence of renal transplantation. In heterozygotes and in a recipient of a renal allograft, elevations and changes in urine glycolipids were less pronounced. In one patient, after several months of ERT, there was a transient increase in Gb3 concentrations to baseline (pre-ERT) levels, associated with the presence of antibodies to the recombinant α-galactosidase A. The marked decline in urine Gb3 on ERT, and its subsequent increase in association with an inhibitory antibody response, suggest that this analyte deserves further investigation as a potential marker of disease severity and response to treatment.
Similar content being viewed by others

References
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917.
Boscaro F, Pieraccini G, la Marca G, et al (2002) Rapid quantitation of globotriaosylceramide in human plasma and urine: a potential application for monitoring enzyme replacement therapy in Anderson-Fabry disease. Rapid Commun Mass Spectrom 16: 1507–1514.
Chaterjee S, Gupta P, Pyeritz RE, Kwiterovich PO (1984) Immunohistochemical localisation of glycosphingolipid in urinary renal tubular cells in Fabry’s disease. Am J Clin Pathol 82: 24–28.
Davies JP, Eng CM, Hill JA, et al (1996) Fabry disease: fourteen alpha-galactosidase A mutations in unrelated families from the United Kingdom and other European countries. Eur J Hum Genet 4: 219–224.
Desnick RJ, Ioannou YA, Eng CM. (2001) α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds; Childs B, Kinzler KW, Vogelstein B, assoc. eds; The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 3733–3774.
Eng CM, Banikazemi M, Gordon RE, et al (2001a) A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance and safety studies. Am J Hum Genet 68: 711–722.
Eng CM, Guffon N, Wilcox WR, et al (2001b) Safety and ecacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med 345: 9–16.
Fujita Y, Mori I, Kitano S (1983) Colour reaction between Pyrogallol Red Molybdenum (VI) complex and protein. Bunseki Kagaku 32: 379–386.
Larsen K (1972) Creatinine assay by a reaction-kinetic approach. Clin Chem Acta 41: 209–217.
Linthorst GE, Hollack CEM, Ormel E, Aerts JMFG (2003) A comparative trial of two enzyme preparations for Fabry disease. From an oral presentation at the 14th ESGLD Workshop 18–21 September 2003, Prodebrady, Czech Republic
MacDermot KD, Holmes A, Miners A (2001) Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 38: 769–777.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalence of lysosomal storage disorders. JAMA 281: 249–254.
Mills K, Johnson A, Winchester B (2002) Synthesis of novel internal standards for the quantitative determination of plasma ceramide trihexoside in Fabry disease by tandem mass spectrometry. FEBS Lett 515: 171–176.
Nakanishi T, Funahashi S, Funai T, Hashimoto T, Akira S (1991) Chemical diagnosis of Fabry’s disease by fluorometric assay and fast atom bombardment/mass spectrometry. Ann Clin Biochem 28: 368–372.
Nelson BC, Roddy T, Araghi S, et al (2004) Globotriaosylceramide isoform profiles in human plasma by liquid chromatography-tandem mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 805: 127–134.
Peters FP, Sommer A, Vermeulen A, Cheriex EC, Kho TL (1997) Fabry’s disease: a multidisciplinary disorder. Postgrad Med J 73: 710–712.
Rosenburg M, Kingma W, Fitzpatrick MA, Richards SM (1999) Immunosurveillance of alglucerase enzyme therapy for Gaucher patients: induction of humoral tolerance in seroconverted patients after repeat administration. Blood 93: 2081–2088.
Schiffmann R, Murray GJ, Treco D, et al (2000) Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA 97: 365–370.
Schiffmann R, Kopp JB, Austin H, et al (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285: 2743–2749.
Wenger DA, Williams C (1991) Screening for lysosomal disorders. In: Hommes FA, ed. Techniques in Diagnostic Human Biochemical Genetics. A Laboratory Manual. New York: Wiley-Liss, 587–617.
Whitfield PD, Sharp PC, Johnson DW, Nelson P, Meikle PJ (2001) Characterization of urinary sulfatides in metachromatic leukodystrophy using electrospray ionization-tandem mass spectrometry. Mol Genet Metab 73: 30–37.
Whitfield PD, Nelson P, Sharp PC, et al (2002) Correlation among genotype, phenotype and biochemical markers in Gaucher disease: implications for the prediction of disease severity. Mol Genet Metab 75: 46–55.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Whitfield, P.D., Calvin, J., Hogg, S. et al. Monitoring enzyme replacement therapy in Fabry disease—Role of urine globotriaosylceramide. J Inherit Metab Dis 28, 21–33 (2005). https://doi.org/10.1007/s10545-005-4415-x
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s10545-005-4415-x