
Abstract
Dengue virus envelope glycoprotein (E-protein) is the main protein associated with immunity induction. To produce a candidate for subunit vaccines and to provide an antigen for diagnostic kits, it was expressed in a novel plant system using deconstructed viral modules. A truncated version of the E-protein was designed to be expressed alone and co-expressed with Dengue virus structural proteins. As well, the critical domain III of E-protein was fused to hepatitis B core antigen (HBcore). The recombinant proteins were produced in Nicotiana benthamiana plants and were reactive with the anti-E antibody. The fusion was reactive with both anti-E and anti-HBcore antibodies.
Similar content being viewed by others


Introduction
Dengue virus (DV) is responsible for causing severe disease and mortality in humans in tropical and subtropical countries. Although the human population at risk of contracting severe dengue related diseases has increased in developing countries, no vaccine against DV is available. Considerable research effort has been made towards developing subunit DV vaccines using different expression systems for the production of DV proteins. Many of them have shown protective immune response from homologous DV challenge in mice (Sugrue et al. 1997; Saejung et al. 2006).
In addition, there is currently a requirement for a cost-effective and safe diagnostic reagent to be used in the detection of DV infections in human population. However, these diagnostic kits are expensive due to the high costs that antigen production represents (AnandaRao et al. 2005).
DV genome encodes three structural proteins C, prM and E (capsid, membrane precursor, and envelope proteins, respectively) and seven non-structural proteins (NS proteins). In host cells, these proteins are translated into a unique polyprotein in which C, prM and E reside in the N-terminal region, while NS proteins reside in C-terminal region. The polyprotein is processed in the endoplasmic reticulum (ER) membrane into individual proteins by both viral and cellular proteases.
E protein has drawn great attention because it is an effective vaccine candidate and a promising antigen for diagnostic kits. E protein is the dominant antigen exposed in the viral surface and contains two N-linked glycosylation sites at Asn-67 and Asn-153. Its responsible for eliciting the first and longest lasting protective antibody response to dengue infection (Bisht et al. 2002). Numerous reports are available describing the production of recombinant DV-E or Et proteins using different expression vectors (e.g. yeast, baculovirus and Escherichia coli). All of them have shown that the recombinant DV-E protein obtained is a suitable immunogen, inducing high titer neutralizing antibodies in mice or rabbits (Bisht et al. 2002; Sugrue et al. 1997; Kelly et al. 2000).
Plant expression system is an alternative platform for the production of recombinant proteins with therapeutic and industrial relevance. Plants as bioreactors retain the advantages of eukaryotic and prokaryotic expression systems currently in use (Twyman et al. 2003; Kumar et al. 2007). However, long production times and low yields of recombinant proteins have been the main bottlenecks in plant-made biopharmaceuticals. A recently developed technique by Icon Genetics (Bayer), known as Magnifection, can offset these disadvantages. The Magnifection system allows very fast production, high recombinant protein expression levels and it is a versatile process that has proved to work successfully with several recombinant proteins (Gleba et al. 2005). For example, the core antigen of the hepatitis B virus (HBcore) was produced by this system in Nicotiana benthamiana with an accumulation level exceeding 7% of total soluble protein (Huang et al. 2006).
To study the potential of plant expression systems as a source for the DV-E immunogenic protein as a future vaccine- and diagnostic kit-component, we explored different expression strategies using the Magnifection system. We designed different expression cassettes to produce a truncated version E (Et), which lacks the membrane anchor domain, the co-expression of Et with DV structural proteins C and prM and a fusion of HBcore with DV serotype 2 domain III of the envelope protein (DV2d3) in N. benthamiana plants.
Materials and methods
Plasmid construction
Vector pMT/V5-HisA C-prM-E/DV carrying the cDNA encoding the DV serotype 2 strain 16681 (GenBank accession number: U87411) structural proteins CprME was given by Dr A. Gamarnik (Instituto Leloir, Argentina). E truncated (Et, aa. 1–397), CprME truncated (CMEt, aa. 1–680) and the fusion of Hepatitis B core antigen (HBcore, aa. 1–154) with the domain III (DVd3, aa. 289–397) of E DV serotype 2 protein (HBcore-DV2d3) were expressed in N. benthamiana plants by generating three different expressions vectors.
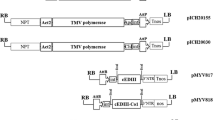
Plasmid SP:Et:K was constructed as follows. Et cDNA was obtained by PCR from pMT/V5-HisA using primers Et-f 5′-AACCCTCCTAATGCGTTGCATAGGA-3′ and Et-r 5′-TCACTA AAGTTCGTCCTTAGAACTTCCTTTCTT-3′, which introduced the KDEL ER retention signal (underlined) and two stop codons in tandem (bold) at 3′-end. Signal Peptide (SP, Glycine-Rich Protein signal peptide) was obtained from the commercial plasmid pCAMBIA1305.2 by PCR using primers SP-f 5′-ATGGCTACTACTAAGCATTT-3′ and SP-r 5′-TGCAACGCATTAGGAGGGTTCTTGC-3′. An overlapping PCR using Platinum Taq DNA polymerase High Fidelity (Invitrogen) was performed to generate SP:Et:K fragment using primers SP-f and Et-r. The resulting PCR fragment was cloned into pGEMT Easy Vector and electropored into DH5α electro-competent cells. Positive clones were screened by PCR and restriction digestions. The EcoRI restriction fragment was ligated into pICH10990 (Giritch et al. 2006) to obtain pSP:Et:K/pICH10990 and subsequently sequenced to assured fidelity to template. The nucleotide and its deduced amino acid sequences were compared with the published sequences for DV-2 strain 16681 in data bases (Fig. 1).

T-DNA schematic representation of ICON constructs used in this study. (Act2: Arabidopsis actin 2 promoter. P-hsp: Arabidopsis heat shock protein promoter. INT: Streptomyces phage PhiC31 integrase. AttP/AttB: PhiC31 integrase recombination sites. RdRp: RNA-dependent RNA polymerase. MP: Movement Protein. 3′NTR: nontranslated region. Nos: Nopaline Synthase terminator. Et: DV Envelope truncated Protein. CMEt: DV Structural proteins. HBc-DV2d3: Fusion HBcore and Dom III of Envelope protein. pICH10990 and pICH11599: ICON vectors.) The expected sizes of each protein product are: Et (45 kDa without glycosylations; 50 kDa glycosylated protein), CMEt (70 kDa) and HBcore-DV2d3 (31 kDa)
In order to obtain CprMEt/pICH10990, CprMEt fragment was obtained by PCR from pMT vector using Platinum Taq DNA polymerase Hi Fidelity using primers CMEt-f 5′-ATGAATAACCAACGGAAAAA-3′ and CMEt-r 5′-TCACTATTGGCCGATAG-3′ which introduced two stop codons (bold) at the 3′-end. The subsequent sub-cloning steps were carried out as described for SP:Et:K/pICH10990.
For HBcore/DV2d3 fusion, the sub-cloning was performed by the ligation of three different restriction fragments. HBcore DNA was obtained from pIBTsHBc (Huang et al. 2006) as an NcoI/KpnI fragment. DNA sequence corresponding to amino acids 289–397 (Domain III, DV E protein) were obtained by PCR from PS:Et:K/pGEMT vector. The primers used were 5′-DV2d3-L-Kpn-f GG GGTACC GGAGGTGGAGGTAGTATGGACAAGCTACAGCTCAA-3′ which introduced a KpnI site for further cloning (underlined) and a Linker peptide (amino acids GTGGGGS, bold) and 5′-DV2d3-6H-Sac-r CCGAGCTCCTAATGGTGATGGTGATGGTGAGAACTTCCTTTCTTAAACCAGTTG-3′ which introduced a hexa-histidine tag (bold), a stop codon and a SacI site (underlined). PCR product was digested with KpnI/SacI. The pICH11599 vector (Marillonnet et al. 2004) was digested with NcoI/SacI. The three different fragments were ligated and the final construct was sequenced to assure fidelity to template. The plasmids were introduced into Agrobacterium tumefaciens GV3101 by electroporation and confirmed by enzyme digestion and PCR after plasmid preparation from transformed bacterial clones.
The 5′-module and the module containing the Integrase were obtained as previously described by Marillonnet et al. (2004).
Agro infiltration of plants
The transformed A. tumefaciens cultures were grown at 28°C for 48 h in YENB medium supplemented with 0.05 mg rifampicin/ml and 0.1 mg carbenicillin/ml for 5′- and Integrase Modules or 0.1 mg carbenicillin/ml for 3′-Module to OD600 of 0.1, 0.15 or 0.2. Cells were harvested by centrifugation at 4,000×g for 10 min at room temperature, resuspended in Infiltration Buffer (10 mM MES, 10 mM MgSO4, pH 5.5) and used at 1:1:1 Integrase Module: 5′ Module: 3′ Module. Leaves of greenhouse-grown N. benthamiana plants, 6–8 weeks old, were infiltrated with the bacterial suspension by applying a syringe without needle in the abaxial side. Infiltrated plants and non-infiltrated control plants (control) were grown in growth chamber at 24°C, 60% humidity and 16/8 h photoperiod until harvesting.
Protein extraction
Infiltrated tobacco leaves were harvested, flash-frozen with liquid N2 and kept at −80°C until the day of the assay. Leaf material (0.1 g fresh weight) was homogenized using two ceramic spheres beads using the FastPrep machine (Fast-prep FP120 Bio, QBiogene) for 45 s at 4.5 m/s in the presence of extraction buffer (1 ml). Three different extraction buffers were tested: Buffer 1 (50 mM sodium phosphate buffer, 100 mM NaCl, 1 mM EDTA, 50 mM sodium ascorbate, 0.01% v/v Triton X-100, 10 μg leupeptin/ml, pH 6,6), Buffer 2 (3% w/v SDS, 1 M DTT, 12.5 mM Tris/HCl, 10% v/v glycerol, 10 μg leupeptin/ml, pH 6.5) and Buffer 3 (2% SDS w/v, 8 M urea, 12.5 mM Tris/HCl, 10% v/v glycerol, 10 μg leupeptin/ml, pH 6.5). The homogenized material was placed on ice for 15 min and centrifuged at 4°C at 8,000×g for 30 min. TSP concentration of extracts was determined by BCA Protein Assay Kit (Pierce) using BSA as the standard.
SDS–PAGE and immunoblot
Proteins were separated by SDS–PAGE using 4–15% (v/v) polyacrylamide Ready Gel (Bio-Rad) and visualized by Coomasie Blue staining. For immunoblot analysis, proteins were transferred to a PVDF membrane and probed with various antibodies. The DV-E protein was detected using a DV proteins antiserum at dilution 1:500 (generous gift of Dr A. Gamarnik, Instituto Leloir, Argentina) followed by reaction with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies (Sigma–Aldrich) diluted 1:3,000. HBcore was probed with an anti-HBcore mouse monoclonal diluted 1:1,000 (ab8638, Abcam) followed by HRP-conjugated goat anti-mouse antibodies (Sigma–Aldrich) at 1:10000. Antibodies were visualized by chemiluminescence using the ECL plus detection reagent (Pierce) and exposed to high performance chemiluminescence film (Amersham Pharmacia).
Results
Et, CMEt and HBcore-DV2d3 transient expression in N. benthamiana leaves
N. benthamiana plants were co-infiltrated with 5′-Module, Integrase Module and three different 3′-Modules carrying coding sequences for Et, CMEt and HBcore-DV2d3 proteins respectively. The different modules were efficiently assembled in planta as we observed efficient recombinant protein production (Fig. 2).

Phenotype of infiltrated N. benthamiana leaves at 5, 7, 8 and 10 days post-infection (dpi) with Et, CMEt and HBcore-DV2d3. The OD600 of infiltration was 0.1. Plant material infiltration was carried out as described in section “Materials and methods”
Three infiltration OD600 were assayed, 0.1, 0.15 and 0.2. As no significant differences between them were found (data not shown), all further experiments were carried out at OD600 = 0.1.
At 5 days post-infection (dpi), CMEt infiltrated leaves showed slight necrosis, while the same phenotype appeared at 7 and 10 dpi for Et and HBcore-DV2d3, respectively. In Et and CMEt expressing leaves, necrosis became more severe as time went on. At 10 dpi, leaves infiltrated with Et and CMEt presented a complete necrosis, resulting in the end point for leaves harvesting (Fig. 2).
Protein extraction
Protein extraction from plant material is a major obstacle. To address this issue, different extraction buffers were assayed. Extraction Buffers 2 and 3 were efficient for protein extraction in Et expressing leaves (Fig. 3a). A band corresponding to the Et plant-produced protein can be seen in lane 3 of SDS–PAGE and the corresponding immunoblot (see black arrows in Fig. 3a). The stronger band at ~50 kDa in SDS–PAGE (lanes 3 and 4) correspond to the molecular mass of RuBisCO large subunit protein. The somewhat higher apparent molecular weight of the protein observed following extraction with buffer 3 is potentially due to the presence of urea in the samples. As was anticipated based on the extent of the necrosis, extraction yielded more recombinant protein at progressively later dpi (Fig. 3b). No specific antigen bands were seen in control leaf extracts. According to these results, we continued the experiments using Extraction Buffer 2.

SDS–PAGE and immunoblot for Et expressing and control (C) leaves. a Protein extraction carried out at 10 days post-infection (dpi) in leaves with Buffer 1, Buffer 2 or Buffer 3. Lanes 1, 2 Et and control leaves extracted with Buffer 1. Lanes 3, 4 Et and control leaves extracted with Buffer 2. Lanes 5, 6 Et and control leaves extracted with Buffer 3. M: Precision Plus Protein Dual Colour Standard (Bio-Rad). b Immunoblot for Et expressing leaves at 6, 7, 8 and 10 dpi extracted with Buffer 2 and Buffer 3. Control extracted with Buffer 2 (Et: PS:Et:K expressed in N. benthamiana leaves. C non-infiltrated control leaves. Black arrows indicate Et protein.)
Et and CMEt plant-produced preliminary characterization
Infiltrated leaf samples were screened at different dpi for Et and CMEt expression by SDS–PAGE and immunoblot (Fig. 4). To normalize the protein amount, loaded volume was adjusted to 30 μg TSP/well. A slight band of ~70 kDa in SDS–PAGE (Fig. 4a) and its corresponding band were obtained for Et and CMEt plant-produced proteins in immunoblot under non-reducing and reducing conditions (Fig. 4b, c respectively).

SDS–PAGE and immunoblot for Et and CMEt produced in N. benthamiana leaves at different days post-infection. a Non-reducing SDS–PAGE. b Immunoblot under non-reducing conditions and c reducing conditions. Immunoblotting and SDS–PAGE were performed as described in section “Materials and methods”. [Et: PS:Et:K/pICH10990 construct. CMEt: CMEt/pICH 10990. C Wt: non-infiltrated control leaves. Black arrow indicates Et or CMEt proteins. M: Precision Plus Protein Dual Colour Standard (Bio-Rad).]
Et and CMEt protein expression was clearly detected at 7, 8 and 10 dpi (Fig. 4b, c). No CMEt protein was detected at 5 dpi. This finding indicates that the recombinant proteins are produced from 7 to 10 dpi at the same levels. The recombinant protein production limiting point is the complete necrosis of the infiltrated leaves.
CMEt protein was expressed and produced successfully with the expected MW. However, higher intensity for Et protein was found. This can be attributed to the protective effect of KDEL retention sequence added to Et recombinant protein. Based on computational programs, the MW for Et is 45 kDa. There is a discrepancy between the theoretical MW and the Et plant-produced MW seen in the SDS–PAGE and immunoblot.
In addition, the immunoblot revealed that several epitopes of Et and CMEt were maintained after the extracts were boiled for 25 min, suggesting that the epitopes of recombinant plant-produced proteins are stable to high temperatures.
HBcore-DV2d3 plant-produced preliminary characterization
Domain III of DV-2 E protein was co-expressed with the HBcore, linked by a flexible glycine-rich linker peptide. The fusion HBcore-DV2d3 was successfully expressed in N. benthamiana leaves. The expected 31 kDa protein was seen at 7, 8, 9 and 10 dpi in SDS–PAGE (Fig. 5a). The corresponding band was also seen in immunoblot when it was probed with rabbit polyclonal anti-DV proteins (Fig. 5b) and mouse monoclonal anti-HBcore antigen antibody (data not shown). No lower bands showed up in either immunoblot for plant-produced fusion protein and no band was detected for control leaves when probed with both antibodies.

SDS-PAGE and immunoblot for the fusion HBcore-DV2d3 produced in N. benthamiana leaves extracted with Buffer 2 at different dpi. a SDS-PAGE for leaves infiltrated with HBcore-DV2d3/pICH11599 at 7, 8, 9 and 10 days post-infection (dpi) and control leaves b Immunoblot for 7, 8, 9 and 10 dpi probed with Rabbit Polyclonal anti-DV proteins serum. Immunoblotting and SDS-PAGE were performed as described in section “Materials and methods”. [Control non-infiltrated control leaves. Black arrow indicates HBcore-DV2d3 fusion protein. M: Precision Plus Protein Dual Colour Standard (Bio-Rad).]
Discussion
In order to produce an efficient antigen, properly folded, and which would preserve the integrity of its neutralizing epitopes, we used a plant expression system capable of producing immunogenic proteins with high yields, guaranteed biosafety, and scalable. We demonstrated that plants represent a valuable alternative eukaryotic expression system for the production of Flavivirus glycoproteins, which can be produced in as short as 7 days using this novel and highly developed plant expression system.
As deleting the carboxy-terminal end of DV serotype 1 (DV-1) E protein could minimize its proteolysis when it was expressed in Pichia pastoris (Bisht et al. 2002), we designed a plant-adapted expression cassette to express this truncated version (Et) in plants. According to computational programs, MW for Et is 45 kDa (without glycosylations). However, Fig. 4 shows that there is a discrepancy between the theoretical MW and the Et plant-produced MW as seen in the SDS–PAGE and immunoblot. This high-molecular weight protein could indicate the presence of a dimeric form of the recombinant protein that could have an electrophoretically different mobility in the gel (45 vs. ~71 kDa). Bisht et al. (2002) observed that when they express the truncated version of DV-2 E protein in P. pastoris, the recombinant protein appeared as high molecular weight aggregates. The aggregates, based on CsCl sedimentation behavior, lacked any ordered structure, indicating that the DV-2 Et protein by itself is unable to form VLPs. They speculated that this type of aggregate can be formed as a result of random intermolecular disulphide bonds forming due to exposure to air oxidation during the protein extraction.
In our study, we took this possibility into account and also considered that a different plant pattern glycosylation (O- and N-glycosylations) could cause differences in gel mobility of plant-produced Et.
Some studies have shown that Flavivirus envelope glycoprotein produced in different expression systems undergoes proteolytic degradation (Wengler et al. 1987; Guirakhoo et al. 1989). In this work, the absence of any discernible low MW protein indicates that there are no plant proteolytic degradation events. We demonstrate that DV-E truncated version with KDEL retention signal or co-expressed with the others structural proteins in plants is not subject to such degradation process. Similar results to ours were obtained by Sugrue et al. (1997a, b) when they expressed the same truncated version of DV-1 in P. pastoris.
The co-expression of DV structural proteins (CprME) has several advantages. Numerous studies suggest that prM protein is required for the proper formation of recombinant E protein antigenic structure (Kelly et al. 2000). DV structural proteins co-expressed in P. pastoris were able to protect the E protein from degradation processes (Sugrue et al. 1997b). On the other hand, the C-terminal region of the C protein has a hydrophobic signal sequence. This signal sequence is needed to anchor the C protein into the ER membrane and, consequently, it allows the translocation of prM and E into the lumen of ER (Qi et al. 2008).
Processing of CprME in mammalian cells depends on two major factors: a particular orientation of the proteins across the ER membrane and the presence of viral and host proteases (Qi et al. 2008). As both factors can be effectively achieved in plant cells, and viral protease is not necessary for prM-E processing, we would have expected the polyprotein to be processed. The anti-serum anti-DV used in our study proved to be able to detect prM protein in previous studies (Talarico and Damonte 2007). As no prM protein (~26 kDa) was detected using this antibody (Fig. 4b, c), we assume that the polyprotein is not being processed in the plant cell, and the ~70 kDa protein represents the unprocessed polyprotein.
Based on our results, no differences were found when expressing Et alone or CMEt in N. benthamiana leaves. However, we cannot guarantee that plants are unable to process the polyprotein successfully because such conclusion would require more investigation.
The three-dimensional structure of DV-E glycoprotein was determined by X-ray crystallography (Modis et al. 2003) and it revealed that the E protein is composed of three domains: Domains I, II and III. Several attempts have been made to express the small and biologically critical Domain III DV. This domain is particularly important for vaccine development as it contains multiple serotype-specific neutralizing epitopes and the host cell receptor recognition site. Dengue virus serotype 2 domain III (DV2d3) has been expressed in N. benthamiana plants (Saejung et al. 2007). DV2d3 produced in plants possessed appropriate antigenicity and immunogenicity and induced neutralizing antibodies in mice.
In this work, DV2d3 was co-expressed with HBcore antigen. HBcore was produced at high-level (7% of TSP or 2.4 mg/g FW) in N. benthamiana leaves using ICON vectors with self-assembly into virus like-particles (Huang et al. 2006). The results indicate the potential use of plant-produced HBcore as a carrier protein for immunogenic presentation of foreign epitopes. In our study, DV2d3 and HBcore antigens were linked by a flexible glycine-rich linker peptide to permit the correct protein folding. Werner et al. (2006) showed that the addition of this kind of flexible linker peptide between the coat protein of turnip vein clearing virus and its A-fusion protein played an essential role in the correct folding of the protein fusion and particle assembly. We expressed the fusion protein in N. benthamiana plants. No lower bands were detected in the immunoblot shown in Fig. 5, indicating that no cleavage occurred in the recombinant fusion protein and that the designed genetic construct was successfully produced.
Protein accumulation levels were estimated from Coomassie-stained gel. We produced Et protein at 0.6 mg/g FW leaf at 7 dpi, CMEt at 0.5 mg/g FW leaf at 10 dpi and HBcore-DV2d3 at 0.4 mg/g FW leaf at 7 dpi.
In conclusion, we have demonstrated that Magnifection is a suitable system for the production of the recombinant DV-2 Envelope protein in N. benthamiana plants. This is the first report of a plant system being able to successfully produce the flaviviral DV-2 E truncated version and its co-expression with C and prM structural DV-2 proteins. Moreover, this is the first work reported in plants in which the HBcore antigen is co-expressed with the critical Domain III of DV-2 envelope glycoprotein. Yield and immunological properties of recombinant proteins are currently being studied.
References
AnandaRao R, Swaminathan S, Fernando S, Jana AM, Khanna N (2005) A custom-designed recombinant multiepitope protein as a dengue diagnostic reagent. Protein Expr Purif 41:136–147
Bisht H, Chugh DA, Raje M, Swaminathan S, Khanna N (2002) Recombinant dengue virus type 2 envelope/hepatitis B surface antigen hybrid protein expressed in Pichia pastoris can function as a bivalent immunogen. J Biotechnol 99:97–110
Gleba Y, Klimyuk V, Marillonnet S (2005) Magnifection—a new platform for expressing recombinant vaccines in plants. Vaccine 23:2042–2048
Guirakhoo F, Heinz FX, Kunz C (1989) Epitope model of tick-borne encephalitis virus envelope glycoprotein E: analysis of structural properties, role of carbohydrate side chain and conformational changes occurring at acidic pH. Virology 169:90–99
Huang Z, Santi L, LePore K, Kilbourne J, Arntzen CJ, Mason HS (2006) Rapid, high-level production of hepatitis B core antigen in plant leaf and its immunogenicity in mice. Vaccine 24:2506–2513
Kelly EP, Greene JJ, King AD, Innis BL (2000) Purified dengue 2 virus envelope glycoprotein aggregates produced by baculovirus are immunogenic in mice. Vaccine 18:2549–2559
Kumar GBS, Ganapathi TR, Srinivas L, Bapat VA (2007) Plant molecular farming: host systems, technology and products. In: Verpoorte R et al (eds) Applications of plant metabolic engineering. Springer, Netherlands, pp 45–77
Marillonnet S, Giritch A, Gils M, Kandzia R, Klimyuk V, Gleba Y (2004) In planta engineering of viral RNA replicons: efficient assembly by recombination of DNA modules delivered by Agrobacterium. PNAS USA 101(18):6852–6857
Modis Y, Ogata S, Clements D, Harrison SC (2003) A ligand-binding pocket in the dengue virus envelope glycoprotein. PNAS USA 100:6986–6991
Qi R, Zhang L, Chi C (2008) Biological characteristics of dengue virus and potential targets for drug design. Acta Biochim Biophys Sin 40(2):91–101
Saejung W, Puttikhunt C, Prommool T, Sojikul P, Tanaka R, Fujiyama K, Malasit P, Seki T (2006) Enhancement of recombinant soluble dengue virus 2 envelope domain III protein production in Escherichia coli trxB and gor double mutant. J Biosci Bioeng 102(4):333–339
Saejung W, Fujiyama K, Takasaki T, Ito M, Hori K, Malasit P, Watanabe Y, Kurane I, Seki T (2007) Production of Dengue 2 envelope domain III in plant using TMV-based vector system. Vaccine 25:6646–6654
Sugrue RJ, Cui T, Xu Q, Fu J, Chan Y (1997a) The production of recombinant dengue virus E protein using Escherichia coli and Pichia pastoris. J Virol Methods 69:159–169
Sugrue RJ, Fu J, Howe J, Chan Y (1997b) Expression of the dengue virus structural protein in Pichia pastoris leads to the generation of virus-like particles. J Gen Virol 78:1861–1866
Talarico LB, Damonte EB (2007) Interference in Dengue virus adsorption and uncoating by carrageenans. Virology 363:473–485
Twyman RM, Stoger E, Schillberg S, Christou P, Fischer R (2003) Molecular farming in plants: host systems and expression technology. Trends Biotechnol 21(12):570–578
Wengler G, Wengler G, Nowak T, Wahn K (1987) Analysis of the influence of proteolytic cleavage on the structural organisation of the surface of the West Nile flavivirus leads to the isolation of a protease-resistant E protein oligomer from the viral surface. Virology 160:210–219
Werner S, Marillonet S, Hause G, Klimyuk V, Gleba Y (2006) Immunoabsorbent nanoparticles based on a tobamovirus displaying protein A. Proc Natl Acad Sci (USA) 103(47):17678–17683
Acknowledgements
CAM is supported by University of Buenos Aires. AMG and JRT are supported by CONICET (National Council of Scientific and Technical Research). CAM, AMG and JRT thank Dr Andrea Gamarnik for her help during the initial stages of this work and Juan A. Mondotte for his assistance. We wish to especially thank Dr Charles J. Arntzen for the opportunity to use the facilities of CIDV (Center for Infectious Diseases and Vaccinology) at the Biodesign Institute to do this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martínez, C.A., Topal, E., Giulietti, A.M. et al. Exploring different strategies to express Dengue virus envelope protein in a plant system. Biotechnol Lett 32, 867–875 (2010). https://doi.org/10.1007/s10529-010-0236-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-010-0236-6