
Abstract
Zerovalent sulfur and inorganic polysulfides were determined in nine sulfide-rich water wells in central and southern Israel. Although the two locations belong to the same aquifer, they are characterized by different pH and hydrogen sulfide levels. Hydrogen sulfide in the central Israel wells ranged between 19 and 32 μM, and the pH was 7.26 ± 0.07. The southern basin is characterized by lower water circulation, lower pH (around 6.8), and higher hydrogen sulfide levels (>470 μM). Polysulfides were determined by a rapid single-phase methylation using methyl trifluoromethanesulfonate (methyl triflate) reagent. The summary polysulfide concentration for S 2−4 –S 2−7 species was found to be around 0.14–0.75 μM in the central region of Israel and substantially higher, 2.3–4.6 μM in the southern region. The sum of polysulfide zerovalent sulfur and colloidal sulfur was quantitatively detected by cyanide derivatization and compared to polysulfide sulfur determined by methyl triflate derivatization and to the chloroform extraction of zerovalent sulfur. A method for the determination of sulfur undersaturation level—the ratio between dissolved elemental sulfur and its equilibrium concentration in the presence of solid sulfur—based on the observed levels of the major polysulfide species is described. The observed polysulfide speciation was compared with the predicted speciation under sulfur saturation conditions taking into account the water temperature, its ionic strength, and pH. Criteria for sulfur saturation versus unsaturated conditions were established based on (1) the chain length dependence of the ratio between the observed polysulfide concentrations and their predicted value under sulfur saturated conditions, and (2) the difference between the concentration of zerovalent sulfur, as determined by cyanolysis, and the total polysulfide sulfur. According to this dual criterion five of the water wells were classified as being undersaturated with respect to sulfur, though for all the examined water wells the majority of the zerovalent sulfur was in the form of polysulfide sulfur.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Despite the hydrophobicity of elemental sulfur, zerovalent sulfur is frequently found in aquifers and water wells. Under anaerobic conditions, the zerovalent dissolved and suspended sulfur may exist in the body of water either as (intracellular or extracellular) colloidal sulfur or as metal-ligated or free polysulfides and hydropolysulfides. Polysulfides are arguably the most intriguing class of compounds among the zerovalent sulfur species in natural aquatic systems. Inorganic polysulfides, S 2− n and their protonated forms are built of linear sulfur chains containing (at least formally) one sulfur atom in a divalent oxidation state and one or more zerovalent sulfur atoms \((\hbox{S}_{n}^{2-}=[\hbox{S}_{n-1}^{(0)}\hbox{S}^{(-2)}]^{2-}).\) This set of compounds, strongly related to the “liver of sulfur” (an aqueous solution of sulfur and potash), was used already by the 9th century Arab (al)chemist Jabir Ibn Hayyan (Geber) (Institute of Arabic and Islamic studies 2007, http://www.islamic-study.org/chemistry.htm), and was investigated more thoroughly by the 18th and 19th century founders of modern chemistry, Scheele (1777), Berzelius (1822), and Mendelejeff (1870). Polysulfides play an important role in numerous environmental processes due to their high reductive and nucleophilic reactivity. Among those processes are transition metal complexation and pyrite formation (Rickard 1975; Howarth 1979; Luther 1991; Paquette and Helz 1997; Chadwell et al. 1999; Luther and Rickard 2005), thiosulfate production by disproportionation to sulfide and thiosulfate under basic conditions (Bloxam 1895; Giggenbach 1974; Licht and Davis 1997), sulfurization of sedimentary organic matter (Aizenshtat et al. 1983; Kohnen et al. 1989; Vairavamurthy and Mopper 1989; Vairavamurthy et al. 1992; Krein and Aizenshtat 1993; Aizenshtat et al. 1995; Amrani and Aizenshtat 2004), volatile sulfur compounds (VSCs) formation (Ginzburg et al. 1999; Heitz et al. 2000; Franzmann et al. 2001; Kamyshny et al. 2003), as well as reductive dehalogenation and nucleophilic substitution of organo-halogen pollutants (Roberts et al. 1992; Perlinger et al. 1996; Miller et al. 1998; Lippa and Roberts 2002; Loch et al. 2002).
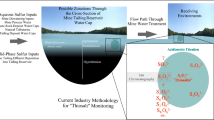
A comprehensive description of reduced sulfur species—with emphasis on zerovalent sulfur—requires the integration of several methods, which are schematically presented in Fig. 1.

Divalent and zerovalent sulfur pools and the relevant methods of analysis
Hydrogen sulfide was quantified by the methylene-blue method (Eaton et al. 1998). This method, which is often referred to as Cline’s method (Cline 1969), relies on the oxidation of N,N-dimethyl-p-phenylenediamine (DPD) by ferric ions and subsequent reaction with acidified hydrogen sulfide samples to give the colored and stable methylene blue. However, the method is not specific for free hydrogen sulfide, and it is rightly referred to as a method for determination of acid labile sulfide (Mylon and Benoit 2001), as it quantifies also other transition metal sulfides (most importantly FeS and MnS) and polysulfides. This method actually sums almost all of the divalent sulfur in the solution including polysulfide and colloid-surface-bound divalent sulfur.
In this work we used the method of derivatization with cyanide (Eq. 1) (Karchmer 1970; Luthy and Bruce 1979) for detection of zerovalent polysulfide sulfur. This method can give a somewhat upwardly biased estimate of the zerovalent polysulfides, because colloidal sulfur can also react with cyanide to give thiocyanate (Eq. 2). Furthermore, polythionates with more than three sulfur atoms also react strongly with cyanide, giving the cyanolysis reaction described by Eq. 3 (Nietzel and De Sesa 1955; Kelly et al. 1969; Szekeres 1974).
The summary level of dissolved and colloidal elemental sulfur concentration was obtained by triple extraction with chloroform and HPLC analysis of the extract (e.g. Goifman et al. 2004a). The method can give an underestimate of the suspended sulfur, since some bioprotected sulfur particles can remain in the aqueous phase even after extraction with organic solvent (Janssen et al. 1999). The method is not specific for elemental sulfur and also reports other zerovalent sulfur forms, particularly zerovalent polysulfide sulfur, which is relevant to the current discussion. Polysulfide sulfur should then be estimated by an alternative method in order to calculate the elemental sulfur.
Various methods were proposed to detect polysulfide speciation under controlled conditions (Maronny 1959; Cloke 1963; Teder 1969; Giggenbach 1972; Boulegue and Michard 1978; Licht et al. 1986; Steudel et al. 1989; Kamyshny et al. 2004). A detailed discussion of the applicability and drawbacks of each of these methods can be found elsewhere (Kamyshny et al. 2004). The detection of polysulfides in natural samples is even more challenging due to their low concentrations, the presence of chemical interferences, and the effects of temperature, pH and the ionic strength on the observed speciation. Polysufide speciation was conducted in this study by methyl triflate derivatization in excess methanol. The method and its validation were recently described (Kamyshny et al. 2004, 2006), and its principle will be only briefly described here. The speciation is based on the rapid, single-phase methylation with methyl trifluoromethanesulfonate, which converts the labile inorganic polysulfides into a stable set of dimethylpolysulfanes (Eq. 4) that can be analyzed by diode array/HPLC.
The key to success of the methyl triflate derivatization, compared to other reported approaches for stabilization by derivatization (Kage et al. 1991; Heitz et al. 2000; Goifman et al. 2004b), is the higher reactivity of the methyl triflate compared to other derivatization agents and the use of excess methanol to increase its miscibility. Luckily, it was found that the methylation step, at least when carried out under specific conditions, is faster than competing reactions that may distort the one-to-one transformation of the polysulfides to their respective polysulfanes. We used a similar method to obtain the enthalpy of polysulfide formation, which facilitates calculation of the temperature-dependent equilibrium distribution of inorganic polysulfides in synthetic solutions (Kamyshny et al. 2007). Both polysulfide chain length and their formation constants by Eq. 5 were found to increase at elevated temperature.
This method was adapted for natural aquatic samples with pH > 6.8, low buffer capacity, and low (submicromolar) concentrations of inorganic polysulfides (Kamyshny et al. 2006) giving the distribution of polysulfides with more than 2 catenated atoms. For lower polysulfides we believe that the methylation under low pH conditions is not fast enough, since these have lower pKa, and their protonated forms are much less reactive compared to their basic forms. In fact, the method was never validated for the disulfide species.
Referring back to Fig. 1, there are three methods for the characterization of the zerovalent sulfur. (1) Derivatization by methyl triflate shows the disproportionation pattern of individual polysulfide species. (2) Extraction with chloroform recovers all particulate sulfur and, partially, polysulfide and colloidal sulfur. (3) Derivatization with cyanide recovers polysulfide sulfur and a portion or even all of the colloidal sulfur. Thus, polysulfide speciation can be given by the triflate method. Suspended sulfur can be roughly estimated by subtracting the calculated sum of zerovalent polysulfide sulfur from the cyanolysis results. Hydrogen sulfide can be estimated from the subtraction of the calculated sum of polysulfides from the methylene-blue observable sulfide. All of those are subject to the assumption that concentrations of organic polysulfanes are low, metal-ligated polysulfides are negligible (we have never proven that these are accounted for by the methylation approach), and that the pH is too high to maintain a significant level of polythionates in the solution. Unfortunately, all the methods involve summation or, even worse, subtraction of experimental observables, which increases significantly the relative analytical error.
2 The Sampled Wells
2.1 Sulfides in Groundwater in Israel
Water wells in Israel exploit groundwater from the uppermost aquifers, in sediments from Quaternary to Cretaceous age. Hydrogen sulfide is formed in groundwater by sulfate reduction, a process which requires the presence of both sulfate and organic matter (which are both usually available) as well as the absence of dissolved oxygen and more potent oxidizers in groundwater. Hydrogen sulfide was found in Israel only in slowly flowing groundwater enclosed in deep aquifers, in their portions that receive very limited recharge. Wells in which H2S was ever found in concentrations exceeding 3 μM (0.1 mg/l) are located mostly in the south (Fig. 2). The maximum H2S concentrations (up to 0.9–1.2 mM) were observed in the deepest wells (>600–700 m), under confined conditions, far from the aquifer outcrops, though even this deep groundwater receives some minor recharge from rain water penetrating at aquifer outcrops. This can be proved by the fact that in several deep wells, where hydrogen sulfide was detected, evidence of periodical O2 appearance is available. This evidence is provided by periodically measured low O2 concentrations in groundwater (Burg et al. 2001, 2003) and by the assemblage of minerals formed in both reducing and oxidizing conditions found in materials clogging the wells (Burg et al. 2003; Zilberbrand et al. 2005). Non-zero tritium concentration (0.7 ± 0.1 TU, Burg et al. 2003) was measured in 1990 in the anoxic confined H2S-bearing groundwater of Nitzana 1 well. This water was pumped out from depths of 672–722 m at a distance of more than 20 km from the outcrops. From the tritium presence it is clear that some recharge arrives to deep groundwater even under such extreme conditions.

Location of sulfide-rich water wells (open circles) and the sampled wells (closed squares)
2.2 Characterization of the Sampled Water Wells
The locations of the wells chosen for this study are marked by full square symbols in Fig. 2. All the wells chosen for sampling (except for Ein Yahav 6) pump groundwater from the Upper-Cretaceous aquifer (Judea Formation), from depths of 98–722 m (Table 1). This aquifer is built of karstic carbonate rock (marine dolomite and calcite) overlaid by slightly permeable marls and chalks of Senonian age. The Ein Yahav 6 water well pumps from depths of 765–878 m, from the Lower-Cretaceous (Kurnub Group) sandy aquifer overlaid by shales. All the wells, except for Kfar Urya, withdraw groundwater from deep, confined portions of the aquifers overlain by low-permeability units.
From the major sulfur compounds, only pyrite (FeS2) was sometimes found in the well cores. Elementary sulfur has never been found in the aquifer, but sometimes it was detected in the sediments clogging the deep wells pumping H2S-bearing water in the northern Negev (Nitzana 3 and Revivim 3—Burg et al. 2003).
The major chemical composition of groundwater in the studied wells was taken from the database of the Hydrological Service of Israel (Table 1). The northernmost wells (Kfar Urya and Mishmar HaNegev) are characterized by a lower salinity and higher pH values. It can be seen that in spite of the H2S presence, all groundwater is strongly enriched in sulfate. The molar SO 2−4 /Cl− ratio exceeds by at least twice the marine value of about 0.0516. There are few data on the fractionation of sulfur isotopes in the relevant wells, but in most cases the isotope enrichment of sulfate relative to hydrogen sulfide is insufficient to conclude that H2S was produced from sulfate in the same aquifer. Values of δ34S in sulfate and in H2S constitute \(2.8{-}6.6\permille\) and \(0.8{-}20.7\permille ,\) respectively, for Nitzana region, whereas in Kfar Urya region they range between 1.3 to 6\(\permille\) and −13.9 to \(-21.4\permille ,\) respectively (Burg et al. 2001, 2003). Sulfate minerals (such as gypsum or anhydrite) were not observed in the vicinity of the sampled wells. Therefore, the enhanced SO 2−4 /Cl− can be evidence of massive oxidation either of solid sulfides (FeS2 and minor minerals associated with it) formed in the past in another environment or of dissolved H2S entering the aquifers via vertical groundwater flow. δ34S values in groundwater sulfate are essentially lower than the values expected in marine sediments (i.e. close to \(15{-}21\permille\) in seawater during Cretaceous-Quaternary period (Clark and Fritz 1997)). This fact points to the influence of the oxidation of isotopically light sulfides. This oxidation can take place at the present, during periods when recharge arrives. However, most of it occurred probably in the past, during the Messinian salinity crisis and Glacial periods, when owing to a strong drop of the level of the Mediterranean Sea the uppermost aquifers were drained and the oxidation zone encroached in formerly water-saturated deep geological layers (Zilberbrand et al. 2005).
The aquifer near the water wells is usually thoroughly protected from penetration of both surface water and groundwater from the upper aquifers. Well casing and cementation of several tens of meters of the well annulus (the space between the casing and the rock formation) prevent the penetration from above. For example, in Nitzana 1 well (Fig. 3), the cemented interval of depths is 259–450 m (within the aquiclude built of clayey chalk and bituminous marl of the Mount-Scopus Group). Above this interval, the previously withdrawn well sediment was used for insulation. Groundwater level in this well was measured at depths of 205–215 and 457–467 m above the well screen (a portion of the casing open to the aquifer). Under such conditions, the entrance of oxygen-bearing water from above through the well casing seems implausible.

Schematic structure of Nitzana 1 well
3 Materials and Methods
3.1 Materials
\(\hbox{Na}_{2}\hbox{HPO}_{4}{\cdot}12\hbox{H}_{2}\hbox{O}\) was from Riedel-de Haën (Seelze, Germany). Ninety-eight percent pure \(\hbox{Na}_{2}\hbox{S}{\cdot}9\hbox{H}_{2}\hbox{O}\) was purchased from Sigma (St. Louis, MO). Elemental sulfur was purchased from Merck (Darmstadt, Germany). \(\hbox{NaH}_{2}\hbox{PO}_{4}{\cdot}\hbox{H}_{2}\hbox{O}\) was purchased from Mallinckrodt (Paris, Kentucky). HPLC grade methanol was purchased from J.T. Baker, Deventer, Holland. Methyl trifluoromethanesulfonate was purchased from Aldrich Chemical Company (Milwakee, WI). Deionized water (<0.2 μS) was used in all tests. Analytical grade reagents were used unless otherwise stated.
3.2 Instruments
Finnigan MAT HPLC system (P4000 pump, AS300 autosampler and UV6000LP detector) was used for separation and detection of dimethylpolysulfanes with Alltech reversed phase C18, 5 μm, 250 mm length, 4.6-mm-diameter column.
3.3 Sampling Methods
The sampling and analysis methods are described in the following sections. Each well was sampled once and one analysis was carried out for each analyte.
3.3.1 Sampling for Detection of Polysulfides by Derivatization with Methyl Triflate
A 100-ml glass bottle was flushed with at least 5 volumes of water, and then a 5-ml sample was immediately taken from the middle height of the bottle with a glass syringe and derivatized in less than 10 s.
3.3.2 Sampling for Detection of Zerovalent Polysulfide Sulfur by Cyanide Derivatization
A 50-ml glass bottle was flushed with at least 5 volumes of water. The sample was derivatized in less than 10 s.
3.3.3 Sampling for Detection of Dissolved and Colloidal Elemental Sulfur by Chloroform Extraction
A 293-ml bottle was flushed with at least 5 volumes of water and closed without any bubble of air inside. Extraction was started within 5 min after sampling.
3.3.4 Sampling for Total Sulfide + Polysulfide S(II-) Measurement
A 100-ml glass bottle was flushed with at least 5 volumes of water, and afterwards 0.5 ml of 1 M solution of ZnCl2 solution and 0.5 ml of 12 M NaOH solution were added immediately for sample conservation. The sample was analyzed in less than 24 h.
3.3.5 Sampling for pH Measurement
A 500-ml glass bottle was flushed with at least 5 volumes of water and closed without any air bubble inside. The pH was measured in less than 12 h after sampling.
3.3.6 Sampling for Sulfate, Sulfite and Thiosulfate Measurement
A 500-ml glass bottle was flushed with at least 5 volumes of water and closed without any air bubble inside. Samples were stored at 4°C in the darkness for 2 weeks before analysis.
3.3.7 Conductivity Measurement
A 500-ml glass bottle was flushed with at least 5 volumes of water and closed without any air bubble inside. Samples were stored at 4°C in the darkness for 2 weeks before the measurement.
3.4 Analytical Methods
3.4.1 Detection of Inorganic Polysulfides by Derivatization with Methyl Triflate
The derivatization procedure was adopted from Kamyshny et al. (2006). The following procedure was used: 40 ml of methanol were vigorously stirred in a septa-closed 100 ml bottle. Three solutions were consecutively injected into the bottle as fast as possible: (1) 5 ml of 50 mM sodium phosphate buffer set to a range of ±0.25 pH units of the sample pH value; (2) 5 ml of the sample; and (3) 200 μl of methyl triflate for samples with pH ≤ 7 or 300 μl of methyl triflate for more basic samples. The derivatized sample was cooled with ice, transferred to the lab, and processed in less than 24 h. Sample was transferred to separatory funnel, 80 ml of 375 mM sodium sulfate solution were added, and the resulting mixture was extracted by 2× 1 ml of n-dodecane. An internal standard (solution of 1,2,5,6-dibenzanthracene in 1,4-dioxane) was added to the n-dodecane extract and 20 μl of the mixture were injected for HPLC analysis. The mobile phase used was 10% water − 90% methanol for 15 min, then a gradient was switched to 100% methanol in 1 min, and 100% methanol was used as a mobile phase for 20 min. Preparation of organic polysulfide standards and HPLC calibration were performed by the method described by Rizkov et al. (2004).
3.4.2 Detection of Zerovalent Polysulfide Sulfur by Derivatization with Cyanide
Fifty milliliters of 0.5% boric acid solution in 250-ml glass were brought to boiling and boiled for 2 min. Fifty milliliters of sample were added to the boiling solution under vigorous stirring, and 0.25 ml of 10% KCN was added at once. The solution was boiled in an open beaker until its volume reached less than 6 ml. After cooling, the solution was brought to 6 ml with water and analyzed for SCN− by ion chromatography. Determination was made under isocratic conditions with an IonPac AS14 anion-exchange column, with 3.5 mM sodium carbonate −1 mM sodium bicarbonate eluent, with a flow rate of 1 ml/min and retention time of 58 min. Maximum standard deviation of the cyanolysis method is 12% for synthetic samples (Kamyshny, in preparation).
3.4.3 Detection of Dissolved and Colloidal Elemental Sulfur by Chloroform Extraction
To 293 ml of the sample 100 μl of internal standard was added and the sample was extracted thrice by chloroform (first extraction—5 ml; second and third extractions—3 ml each) in a 500 ml separatory funnel. All extracts were united and the resulting solution was analyzed by HPLC under isocratic conditions (using pure methanol as the mobile phase). Maximum standard deviation of the chloroform extraction method is 8% for synthetic samples (Kamyshny, in preparation).
3.4.4 Total Sulfide + Polysulfide S(II) Measurement
Hydrogen sulfide was determined according to the Eaton et al. (1998) protocol using the 4500-S2- D procedure.
3.4.5 Conductivity Measurement
Conductivity was measured in the untreated sample. Ionic strength was calculated by the following, mixed units, empirical formula (Snoeyink and Jenkins 1980):
4 Results
Polysulfide speciation by methyl trifluoromethanesulfonate allowed us to detect polysulfides with 4–7 sulfur atom chains (columns 7–10 in Table 2). A typical chromatogram of polysulfane speciation after methyltriflate derivatization is depicted in Fig. 4. The peaks in the chromatogram are wide which allows a relatively large amount of possible interferences.

Typical HPLC chromatogram for the Tzofar 220 analysis of dimethylpolysulfanes
To estimate the errors in the detected concentrations of polysulfides two sources of error were taken into account. (1) The reported relative standard deviation (RSD) of determination of each of the polysulfides: 14, 7, 9, and 21% for S 2−4 , S 2−5 , S 2−6 and S 2−7 , respectively (Kamyshny et al. 2006). These values were considerably higher than the values found for the RSDs of nitrogen purged and then polysulfide spiked Tzofar 220 water. The relative standard deviations of (seven measurements of) spiked polysulfides in Tzofar 220 were 3.3, 2.0, 5.6, and 13.1% for the tetra- to hepta-sulfide species respectively. (2) The background noise in the integrated areas. The latter was estimated by integration of the background noise per 1 min interval multiplied by average width of each peak. The relative noise was calculated by dividing the obtained figure by the different peak areas. These two relative errors were summed up to produce the data in Fig. 5 and Table 2. Table 2 depicts also the measured pH and electrical conductivity. The ionic strength was calculated using Eq. 6, and the results are depicted in column 3 of Table 2. The concentration of zerovalent sulfur in dissolved state bound in polysulfides can be calculated by Eq. 7. These concentrations are depicted in Table 3 (in the column titled S°–A).

Observed tetrasulfide (blank bars), pentasulfide (vertical lines), and hexasulfide (dotted bars). Error bars refer to the calculated error of polysulfide species detection (see text for details)
The difference between groundwater in central and southern Israel can be easily discerned from Table 2. The southern aquifers wells have higher total dissolved solids (and conductivity), lower pH, and higher hydrogen sulfide levels. Although a decrease in pH and an increase in hydrogen sulfide influence the equilibrium level of polysulfides in opposite directions, it is clear that in our cases the influence of the higher hydrogen sulfide levels in the southern aquifers dominates over the influence of their lower pH, and the polysulfide levels are much higher in southern Israel compared to central Israel.
The experimental data on concentrations of individual polysulfides were compared with the values calculated from temperature-dependant equilibrium constants derived by Kamyshny et al. (2007), using the assumption that the sample is supersaturated with respect to sulfur and taking into account the temperature, pH, and ionic strength. The latter was calculated using Davies equation (Stumm and Morgan 1996)
where f is the activity coefficient, z the charge, and I is the ionic strength.
Every concentration value in Table 2 is accompanied by recovery percent values (in brackets) calculated by Eq. 9.
where C exp is the experimentally determined concentration of polysulfide species and C calc the concentration of polysulfide species calculated using thermodynamic constants from Kamyshny et al. (2007). The data related to the estimation of the zerovalent sulfur by cyanolysis and chlorform extraction are depicted in Table 3 (columns 3 and 4).
5 Discussion
5.1 Water Properties
The targeted wells can be divided into two groups according to the concentrations of major ions in groundwater (Table 1) as well as their polysulfide and zerovalent sulfur concentrations (Tables 2 and 3). In three wells in central Israel, Kfar Urya 2, Kfar Urya 8, and Mishmar HaNegev 3, the hydrogen sulfide concentration is relatively low (19–32 μM), and the pH is slightly basic (7.29–7.42), while the salinity and the electrical conductivity are rather high. The second group consists of the six southern wells (Nitzana 1, Ein Yahav 6, Tzofar 220, Tzofar 221, Paran 18, and Paran 19). Here, hydrogen sulfide is relatively high (470–1010 μM), the pH is slightly acidic (6.79–6.88), and the water is brackish. Kfar Urya 2 and Kfar Urya 8 wells have relatively low thiosulfate concentrations (<10 μM), whereas in the southern wells the concentration of thiosulfate was in the range of 11–61 μM—notably higher than zero.
5.2 Polysulfide Speciation
The speciation of the polysulfides is graphically depicted in Fig. 5 for all the nine wells. A detailed account of the speciation equations was described by Kamyshny et al. (2004). Briefly, it is useful to relate to polysulfide formation from dissolved elemental sulfur and hydrogen sulfide (and the related formation constant).
where {S} denotes the activity of dissolved elemental sulfur. When saturated conditions prevail, it is simpler to use the formation constant from solid sulfur (activity = 1), which simply gives:
Equation 11 is the basic working tool for speciation calculations, though it relates only to sulfur saturated conditions. Speciation by Eq. 11 does not require prior knowledge of the sulfur dissolution constant or its equilibrium dissolved activity level, {S}s. K n values and, at times, their temperature dependencies, were derived by several authors, and a summary of these values along with our predicted thermodynamic constants can be found in Kamyshny et al. (2007). In agreement with the predictions of Kamyshny et al. (2007), the pentasulfide species is the dominant species, though in practically all cases the hexasulfide was one of the three dominant polysulfides (Fig. 5). The actual recovery compared to the thermodynamic predictions (assuming sulfur saturated conditions) ranged between 26 and 141% for the three dominant polysulfide species (recovery values are depicted in brackets in Table 2). Heptasulfide was also found in significant quantities, but its level was usually low as compared to the three dominant species, and it was below detection limit for all of the central-basin wells (i.e., the Kfar Urya wells and Mishmar HaNegev). Under the close-to-neutral pH conditions of the inspected wells, the level of disulfide is less than 1% of its hydrodisulfide form, and since the latter is of much lower nucleophilicity, the disulfide species do not quantitatively convert to the respective dimethyldisulfane upon methyl triflate addition, and they were left unquantified in the current study.
The groundwater temperature in the different wells ranged between 27 and 44°C. This 17° difference has a significant effect on the calculated equilibrium of different polysulfides. In order to visualize the temperature dependence of speciation, we present the equilibrium distribution of tetra- to hepta-sulfides in Fig. 6 for the two extreme temperature conditions (27°C is presented by solid lines and 44°C by dashed lines). The ionic strength has little effect on speciation, and it was kept constant in these calculations. The speciation was calculated for total divalent sulfur of 1 M, but the same relative distribution of divalent sulfides holds for any other total divalent sulfur concentration (provided of course that sulfur saturated conditions prevail). All the constants required for the temperature-dependent calculation of the speciation are available (Kamyshny et al. 2007), except for the enthalpy of dissociation of the polysulfanes and the hydropolysulfanes. The important paper of Schwarzenbach and Fischer (1960) derives the pKa’s of the polysulfanes only at 25°C.

Equilibrium distribution of hydrogen sulfide (upper four curves, corresponding to HS− and H2S at two temperatures) and S 2−4 (blue), S 2−5 (green), S 2−6 (orange), and S 2−7 (purple) under sulfur saturated conditions for 27° (solid line) and 44°C (dashed lines). Coloured data points depict the observed polysulfide levels in this study. The colors of the different polysulfide data points are identical to the respective lines. 1, Paran 18 and Ein Yahav 6; 2, Tzofar 220 and Tzofar 221; 3, Nitzana 1; 4, Paran 19; 5, Mishmar HaNegev—3; 6, Kfar Urya—8; 7, Kfar Urya—2
Figure 6 shows that the order of relative significance of the polysulfides is maintained at the range of temperatures and pH levels encountered in this research: Pentasulfide being the most dominant polysulfide species is followed by tetrasulfide and hexasulfide, which have approximately the same concentrations. The different data points on Fig. 6 mark the observed distribution of polysulfides normalized to 1 M total divalent sulfur (i.e. we divided each concentration by the concentration of the total divalent sulfur found in the well). The qualitative agreement between the observations and the model predictions is apparent.
Pentasulfide was the most abundant species in seven of the nine tested wells, and it may serve as a good predictor for the overall sum of zerovalent polysulfide sulfur. Columns 2 and 5 of Table 3 depict the observed and the calculated sum of polysulfides, respectively. The calculations are based on the observed level of pentasulfide (Eq. 12).
c K denotes the different equilibrium constants for infinite dilution; Ka 1,n and Ka 2,n correspond to the first and second deprotonation constants of the different polysulfanes. Indeed, the observed and calculated levels correlated very well, as depicted in the linear correlation of Fig. 7a. The only significant deviation was found for the Mishmar HaNegev well, where the observed level of polysulfides was the lowest, and for which only pentasulfide exceeded the minimum detection level.

(a) A correlation between the calculated level of zerovalent polysulfide sulfur (from Eq. 12) and its observed level. (b) Graphic presentation of the unsaturation criterion of Eq. 19 for Kfar Urya 2 (closed circles and trendline 1); Paran 18 (closed diamonds and trendline 2); Ein Yahav 6 (closed rectangles and trendline 3); Tzofar 221 (closed triangles and trendline 4); Paran 19 (open circles and trendline 5); Nitzana 1 (open circles and trendline 6); Kfar Urya 8 (open rectangles) and Tzofar 220 (open diamonds)
5.3 Sulfur Undersaturation Versus Saturation Conditions
Elemental sulfur solubility is so low, and the overlapping contribution of different zerovalent sulfur sources to the observable zerovalent sulfur (Fig. 1) is so high, that the distinction between sulfur saturation conditions and undersaturation conditions is challenging. Polysulfide speciation provides an interesting way to identify undersaturation. Dividing the equilibrium constant for the formation of S 2−n+m (Eq. 10) by the formation constant of S 2− n gives Eq. 13
The relationship between the two equilibrium constants is given by Eq. 14.
where {S}s is the solubility of elemental sulfur, which usually relates to the orthorhombic phase (note that {S}s ≠1, it is not the activity of the solid). Substitution of Eq. 14 in Eq. 13 gives
It is useful to define the “relative saturation level” (RSL) as the ratio between the actual activity of dissolved elemental sulfur {S} and its activity under saturated conditions {S}s
Using the newly defined RSL value, the relative concentration of polysulfide species is given by Eq. 17
The ratio between the relative concentrations of polysulfide of n + m and n sulfur atoms at undersaturation \(\left[\left\{\hbox{S}_{n+m}^{2-} \right\}/\left\{\hbox{S}_n^{2-}\right\}\right]\) and saturation \(\left[ \left\{\hbox{S}_{n+m}^{2-}\right\}/\left\{\hbox{S}_n^{2-}\right\} \right]_{\rm s}\) is given by dividing both sides of Eq. 17 for under-saturation (RSL < 1) by the relevant terms of Eq. 17 for saturation (RSL = 1) conditions:
Equation 18 implies that the actual speciation—at undersaturation conditions—deviates from the calculated speciation under saturation conditions, and that the degree of deviation is a simple function of the RSL and the number of sulfur atoms in the polysulfide. After rearrangement (and assigning n = 5), it is possible to get a linear relationship, in which (n−5) is the independent variable and \(\log\left[\left\{\hbox{S}_n^{2-}\right\}/\left\{\hbox{S}_n^{2-}\right\}_{\rm s} \right]\) is the dependent variable, with a slope equal to log(RSL).
Equation 19 can be used to calculate the speciation when the RSL is known, and more relevant to the current study, it can be used to calculate the RSL itself, and thus serves as a criterion for undersaturation conditions, provided that polysulfide speciation is known. The polysulfide activities of Eq. 19 can be replaced by the sum of the relevant polysulfides, hydropolysulfides, and polysulfanes, when the pH is close to the relevant pKa’s of the polysulfanes (i.e. \(\alpha _2 \Upsigma S_{n}^{2-}\equiv\alpha_2 ([\hbox{S}_n^{2-}]+[\hbox{HS}_n^-]+[\hbox{H}_2 \hbox{S}_n])=[\hbox{S}_n^{2-}],\) where α2 is the second ionization fraction of the polysulfane). However, due to the acidity of the high hydropolysulfides, the use of ionization fractions is not necessary for the tetrasulfide and higher polysulfides under near-neutral and basic conditions.
Figure 7b depicts the use of Eq. 19, for eight groundwater wells for which polysulfide speciation was available. The figure shows that six of the eight groundwater wells obey the linear dependence of Eq. 19 with RSL < 0.8 and correlation coefficient, R 2 > 0.8. The right column of Table 3 delineates the obtained RSLs and the associated error in the determination of the slopes of the curve in Fig. 7b. The estimated errors in the determination of the concentrations of polysulfides (Table 2) were used to calculate the maximum RSL errors. The lowest estimate of an RSL was determined by fitting Eq. 19 (i.e. drawing a line similar to those depicted in Fig. 7b) using the level of S 2−4 plus the error in its determination and the level of S 2−6 minus its estimated error. An estimate of the upper level of RSL for each well was gained by fitting Eq. 19 with the minimal value of S 2−4 and the maximal value of S 2−6 . The RSLs were not very sensitive to the rather large error margins in the determination of the different concentrations of the polysulfides, and except for the Kfar Urya 2 well the error in slope was less than 0.23 (in absolute values). This relative insensitivity is associated with the logarithmic form of Eq. 19. We also calculated the standard deviation of the slopes of Fig. 7b by a straightforward regression analysis and by the method of weighted regression analysis (Miller and Miller 1993). In both cases the relative error was less than 3.5% which we consider negligibly small and therefore these error estimates were not presented.
Another criterion which supports our conclusions regarding the undersaturation of most of the targeted water wells is the fact that zerovalent sulfur by cyanolysis (column 4 in Table 3) is lower than or exceeds by less than 10% the polysulfide sulfur estimate (column 6 of Table 3). In Paran 18 and Tzofar 220 the two criteria give contradicting predictions (though in opposite directions). Thus it can be safely stated that undersaturated conditions prevail in Kfar Urya 2, Nitzana 1, Ein Yahav 6, Tzofar 221, and Paran 19, whereas sulfur saturation prevails in Kfar Urya 8 and the situation is unclear for Mishmar-HaNegev-3, Tzofar 220, and Paran 18.
5.4 Analysis of the Zerovalent Sulfur Pools
Zerovalent sulfur is estimated in this study by three independent methods: chloroform extraction, cyanolysis, and by summing the polysulfide sulfur contributions. As could be expected, we did not find a one-to-one correspondence between the different measures of zerovalent sulfur, but surprisingly, the correlations between the three estimates for zerovalent sulfur were very low. Chloroform extraction is the method of choice when it comes to quantification of total zerovalent sulfur in surface waters and sediments, since it takes into account the solid sulfur pools. However, when it comes to deep groundwater, the precipitate contribution is very low. Indeed the correlation between the chloroform extraction results (S°-B) and the other two methods is very low (R 2 < 0.4). The correlation between cyanolysis and polysulfide sulfur pool is somewhat higher (R 2 = 0.69), because the polysulfide sulfur in the studied wells is a major contributor to both methods (and we suspect that this holds true for most water wells due to the filtration of the macro-size zerovalent sulfur fraction by the soil).
5.5 Prediction of Disulfide Level
Disulfide level is probably the most disputed variable in the still argued polysulfide speciation. Figure 8 depicts the calculated relative contributions of zerovalent polysulfide sulfur as a function of the pH level calculated based on different thermodynamic sets. For the high pH end, the contribution of disulfide and its protonated forms is unarguably negligible. The situation is different for the near-neutral pH levels. Here, Maronny’s and our predictions far surpass others’ predictions. Unfortunately, our K 2 values are based on high-pH derivatizations, and since under such conditions the disulfide levels were very low, it is perfectly possible that our error margins are somewhat higher than originally expected. Our high K 2 value influences also our estimate for the zerovalent polysulfide sulfur based on the sulfur saturation assumption as depicted in Table 3 (right column). The predictions based on polysulfide saturation, which take into account disulfide bound zerovalent sulfur, are at times 50% higher than the predictions based on negligible disulfide contribution.

Percent of zero-valent polysulfide sulfur in all disulfide species (H2S2, HS2−, and S 2−2 ) as a function of pH at standard conditions, calculated from literature data. 1, Kamyshny et al. (2007); 2, Maronny (1959); 3, Giggenbach (1972); 4, Boulegue (1978); 5, Cloke (1963). The shaded rectangle depicts the pH range of the studied wells. The proton dissociation constants (Ka‘s) of the polysulfanes were taken from Schwarzenbach and Fischer (1960)
5.6 Oxidation Source
An important enigma, which accompanies our research and for which we have no satisfactory answer, relates to the source of oxidizing agent in the studied aquifers. Formation of sulfur and, even more so, thiosulfate requires penetration of oxygen or mixing of the groundwater with another source of oxygen-rich water (though nitrate is also capable of bio-oxidizing the hydrogen sulfide, and FeO(OH) or MnO2 can oxidize hydrogen sulfide chemically as well as microbiologically). Polysulfides are formed in oxidation processes. Thus, the presence of polysulfides in the studied wells agrees with the hypothesis related to the external/ancient source of hydrogen sulfide and the sporadic appearance of dissolved oxygen in the aquifer (Zilberbrand et al. 2005). Another explanation for the presence of the polysulfides could be in their production by oxidation in the well pipe itself. As it was mentioned above, this explanation is unlikely owing to well insulation from the land surface and the high water column in the well. In addition, all groundwater samples were taken after intensive, at least 1-h pumping. That is, even if some small flux of oxygen-bearing water from upper aquifers could enter through the borehole casing, it should be incomparably smaller than the lateral groundwater flux. Considering the kinetics of oxidation processes, the portion of polysulfides formed in the borehole in total detected polysulfides should be very small.
6 Concluding Remarks
Zerovalent sulfur was determined in this study under the most challenging conditions of low pH and very low sedimentary sulfur conditions. Under such conditions, polysulfides serve as the main pool of zerovalent sulfur and their inherent chemistry determines the level of the observed zerovalent sulfur. High pH and hydrogen sulfide levels increase polysulfide concentrations and zerovalent sulfur abundance.
In this article we presented a dual criterion for sulfur undersaturation based on polysulfide speciation and the ratio between the levels of observable zerovalent sulfur detected by cyanolysis and the predicted polysulfide-sulfur pool. When the RSL calculated based on Eq. 19 is significantly lower than 1 (we used a value of 0.8 in the current article), and additionally, the ratio between the zerovalent sulfur detected by cyanolysis and the zerovalent sulfur estimated by polysulfide derivatization is close (or lower) to unity, one can assume that undersaturation with respect to sulfur prevails. Naturally, in such cases, polysulfides are the only significant zerovalent sulfur pool.
References
Aizenshtat Z, Stoler A, Cohen Y, Nielsen H (1983) The geochemical sulphur enrichment of recent organic matter by polysulfides in the Solar Lake. In: Bjøroy M (ed) Advances in organic geochemistry, Proc. 10th Int. Meet. 1981. Wiley, Chichester, pp 279–288
Aizenshtat Z, Krein E, Vairavamurthy M, Goldstein T (1995) Role of sulfur in the transformation of sedimentary organic matter: a mechanistic overview. In: Vairavamurthy A, Schoonen MAA (eds) Geochemical transformations of sedimentary sulfur. American Chemical Society, Washington, DC, pp 16–37
Amrani A, Aizenshtat Z (2004) Reaction of polysulfide anions with, alpha, beta-unsaturated isoprenoid aldehydes in aquatic media: simulation of oceanic conditions. Org Geochem 35:909–921
Berzelius J (1822) De la Composition des Sulfures Alcalins. Ann Chim Phys 20:113–141
Bloxam WP (1895) The sulphides and polysulphides of ammonium. J Chem Soc Trans 67:277–309
Boulegue J (1978) Metastable sulfur species and trace metals (Mn, Fe, Cu, Zn, Cd, Pb) in hot brines from the French Dogger. Am J Sci 278:1394–1411
Boulegue J, Michard G (1978) Constantes de formation des ions polysulfures S 2−6 , S 2−5 et S 2−4 en phase aqueuese. J Franç Hydrol 9:27–33
Burg A, Gavrieli Y, Guttman J (2001) Factors for development of reductive conditions in Judea-Group aquifer within Judea Lowlands and recommendations for exploitation. Report of the Israeli Geological Survey GSI/9/2001, Jerusalem, 20 pp (in Hebrew)
Burg A, Gavrieli Y, Guttman J (2003) Hydrogeology and hydrogeochemistry of a brackish groundwater in the south of the Be’er Sheba basin and their connection to water-well clogging. Report of the Israeli Geological Survey GSI/35/2003, Jerusalem, p 33 (in Hebrew)
Chadwell SJ, Rickard D, Luther GW III (1999) Electrochemical evidence for pentasulfide complexes with Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+. Aquat Geochem 5:29–57
Clark ID, Fritz P (1997) Environmental isotopes in hydrogeology. New York, Lewis Publishers, p 328
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Cloke PL (1963) The geologic role of polysulfides. 1. The distribution of ionic species in aqueous sodium polysulfide solutions. Geochim Cosmochim Acta 21:1265–1298
Eaton AD, Clesceri LS, Greenberg AE (1998) Standard methods for the analysis of water and wastewater. APHA, Washington
Franzmann PD, Heitz A, Zappia LR, Wajon JE, Xanthis K (2001) The formation of malodorous dimethyloligosulfides in treated groundwater: The role of biofilms and potential precursors. Water Res 35:1730–1738
Giggenbach W (1972) Optical spectra and equlibrium distribution of polysulfide ions in aqueous solutions at 20°C. Inorg Chem 11:1201–1207
Giggenbach W (1974) Equilibria involving polysulfide ions in aquous solution up to 240°. Inorg Chem 13:1724–1730
Ginzburg B, Dor I, Chalifa I, Hadas O, Lev O (1999) Formation of dimethyloligosulfides in lake Kinneret: biogenic formation of inorganic oligosulfide intermediates under oxic conditions. Environ Sci Technol 33:571–579
Goifman A, Rizkov D, Gun J, Kamyshny A Jr, Modestov AD, Lev O (2004a) Inorganic polysulfides quantitation by methyliodide derivatization: dimethylpolysulfide formation potential. Water Sci Technol 49:179–184
Goifman A, Gun J, Gitis V, Kamyshny A Jr, Lev O, Donner J, Börnick H, Worch E (2004b) Pyrolysed carbon supported cobalt porphyrin: a potent catalyst for oxidation of hydrogen sulfide. Appl Catal B Environ 54:225–235
Heitz A, Kagi RI, Alexander R (2000) Polysulfide sulfur in pipewall biofilms: its role in the formation of swampy odour in distribution systems. Water Sci Technol 41:271–278
Howarth RW (1979) Pyrite: its rapid formation in a salt marsh and its importance in ecosystem metabolism. Science 203:49–51
Institute of Arabic and Islamic Studies (2007). http://www.islamabad.net/science.htm
Janssen AJH, Lettinga G, de Keizer A (1999) Removal of hydrogen sulphide from wastewater and waste gases by biological conversion to elemental sulphur – colloidal and interfacial aspects of biologically produced sulphur particles. Colloids Surf A Physicochem Eng Asp 151:389–397
Kage S, Nagata T, Kudo K (1991) Determination of polysulfides in blood by gas chromatography – mass spectrometry. J Chromatogr 564:163–169
Kamyshny A Jr, Goifman A, Rizkov D, Lev O (2003) Formation of carbonyl sulfide by the reaction of carbon monoxide and inorganic polysulfides. Environ Sci Technol 37:1865–1872
Kamyshny A Jr, Goifman A, Gun J, Rizkov D, Lev O (2004) Equilibrium distribution of polysulfide ions in aqueous solutions at 25°C: a new approach for the study of polysulfides’ equilibria. Environ Sci Technol 38:6633–6644
Kamyshny A Jr, Ekeltchik I, Gun J, Lev O (2006) A method for the determination of inorganic polysulfide distribution in aquatic systems. Anal Chem 78:2631–2639
Kamyshny A Jr, Gun J, Rizkov D, Voitsekovski T, Lev O (2007) Equilibrium distribution of polysulfide ions in aqueous solutions at different temperatures by rapid single phase derivatization. Environ Sci Technol 41:2395–2400
Karchmer JK (1970) Analytical chemistry of sulfur and its compounds. Wiley-Interscience, New York, p 349
Kelly DL, Chambers LA, Trudinger PA (1969) Cyanolysis and spectrophotometric estimation of trithionate in mixture with thiosulfate and tetrathionate. Anal Chem 41:898–901
Kohnen MEL, Sinninghe Damsté JS, ten Haven HL, de Leeuw JW (1989) Early incorporation of polysulfides in sedimentary organic matter. Nature 341:640–641
Krein EB, Aizenshtat Z (1993) Phase transfer-catalyzed reactions between polysulfide anions and α,β-unsaturated carbonyl compounds. J Org Chem 58:6103–6108
Licht S, Davis J (1997) Disproportionation of aqueous sulfur and sulfide: kinetics of polysulfide decomposition. J Phys Chem B 101:2540–2545
Licht S, Hodes G, Manassen J (1986) Numerical analysis of aqueous polysulfide solutions and its applications to cadmium chalcogenide polysulfide photoelectrochemical solar cells. Inorg Chem 25:2486–2489
Lippa KA, Roberts AL (2002) Nucleophilic aromatic substitution reactions of chloroazines with bisulfide (HS−) and polysulfides (S 2−n ). Environ Sci Technol 36:2008–2018
Loch AR, Lippa KA, Carlson DL, Chin YP, Traina SJ, Roberts AL (2002) Nucleophilic aliphatic substitution reactions of propachlor, alachlor and metolachlor with bisulfide (HS−) and polysulfides (S 2−n ). Environ Sci Technol 36:4065–4073
Luther GW III (1991) Pyrite synthesis via polysulfide compounds. Geochim Cosmochim Acta 55:2839–2849
Luther GW, Rickard DT (2005) Metal sulfide cluster complexes and their biogeochemical importance in the environment. J Nanopart Res 7:389–407
Luthy RG, Bruce SG Jr (1979) Kinetics of reaction of cyanide and reduced sulfur species in aqueous solutions. Environ Sci Technol 13:1481–1487
Maronny G (1959) Constantes de dissotiation de l‘hydrogene sulfure. Electrochim Acta 1:58–69
Mendelejeff DI (1870) Constitution der Polythionsäuren. Berichte der Deutschen Chemischen Gesellschaft 3:870–873
Miller JC, Miller JN (1993) Statistics for analytical chemistry, 3rd edn. Ellis Horwood Ltd., New York
Miller PL, Vasudevan D, Gschwend PM, Roberts AL (1998) Transformation of hexachloroethane in a sulfidic natural water. Environ Sci Technol 32:1269–1275
Mylon SE, Benoit G (2001) Subnanomolar detection of acid labile sulfides by the classical methylene blue method coupled to HPLC. Environ Sci Technol 35:4544–4548
Nietzel OA, De Sesa MA (1955) Spectrophotometric determination of tetrathionate. Anal Chem 27:1839–1841
Paquette KE, Helz GR (1997) Inorganic speciation of mercury in sulfidic waters: the importance of zero-valent sulfur. Environ Sci Technol 31:2148–2153
Perlinger JA, Angst W, Schwarzenbach EP (1996) Kinetics of the reduction of hexachloroethane by juglone in solutions containing hydrogen sulfide. Environ Sci Technol 30:3408–3417
Rickard DT (1975) Kinetics and mechanism of pyrite formation at low temperatures. Am J Sci 275:636–652
Rizkov D, Lev O, Gun J, Anisimov B, Kuselman I (2004) Development of in-house reference materials for determination of inorganic polysulfides in water. Accredit Qual Assur 9:399–403
Roberts AL, Sanborn PN, Gschwend PM (1992) Nucleophilic-substitution reactions of dihalomethanes with hydrogen-sulfide species. Environ Sci Technol 26:2263–2274
Rozan TF, Lassman ME, Ridge DP, Luther GW III (2000) Evidence for iron, copper and zinc complexation as multinuclear sulphide clusters in oxic rivers. Nature 406:879–882
Scheele C (1777) Chemische Abhandlung von der Luft und dem Feuer. Upsala-Leipzig, p 153
Schwarzenbach G, Fischer A (1960) Die Acidität der Sulfane und die Zusammensetzung wässeriger Polysulfidlösungen. Helvetica Chimica Acta 43:1365–1390
Snoeyink VL, Jenkins D (1980) Water chemistry. Wiley, New York
Steudel R, Holdt G, Göbel T (1989) Ion-pair chromatographic separation of inorganic sulphur anions including polysulphide. J Chromatogr 475:442–446
Stumm W, Morgan JJ (1996) Aquatic chemistry, 3rd edn. Wiley, New York
Szekeres L (1974) Analytical chemistry of the sulphur acids. Talanta 21:1–44
Teder A (1969) The spectra of aqueous polysulfide solutions. Part II: the effect of alkalinity and stoichiometric composition at equilibrium. Arkiv För Kemi 31:173–198
Vairavamurthy A, Mopper K (1989) Mechanistic studies of organosulfur (thiol) formation in coastal marine sediments. In: Biogenic sulfur in the environment. American Chemical Society, Washington, DC, pp 231–242
Vairavamurthy A, Mopper K, Tailor BF (1992) Occurrence of particle-bound polysulfides and significance of their reaction with organic matters in marine sediments. Geophys Res Lett 19:2043–2046
Zilberbrand M, Rosenthal E, Guttman J, Weinberger G, Friedman V (2005) Updating the conceptual model of the Yarkon-Tanninim basin: boundaries, hydrogeochemistry and salinization processes. Report of the Hydrological Service of Israel, Hydro/1/2005, Jerusalem, p 49 (in Hebrew)
Acknowledgements
The authors are grateful for the financial support of the Water Technology Program of the MOS, Israel, and BMBF, Germany, the financial support by Mekorot Water Company Ltd. A.K. thanks MPG Minerva Post-Doctoral Fellowship for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kamyshny, A., Zilberbrand, M., Ekeltchik, I. et al. Speciation of Polysulfides and Zerovalent Sulfur in Sulfide-rich Water Wells in Southern and Central Israel. Aquat Geochem 14, 171–192 (2008). https://doi.org/10.1007/s10498-008-9031-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10498-008-9031-6