
Abstract
It is generally acknowledged that driver pathway plays a decisive role in the occurrence and progress of tumors, and the identification of driver pathways has become imperative for precision medicine or personalized medicine. Due to the inevitable sequencing error, the noise contained in single omics cancer data usually plays a negative effect on identification. It is a feasible approach to take advantage of multi-omics cancer data rather than a single one now that large amounts of multi-omics cancer data have become available. The identification of driver pathways by integrating multi-omics cancer data has attracted attention of researchers in bioinformatics recently. In this paper, a weighted non-binary mutation matrix is constructed by integrating copy number variations, somatic mutations and gene expressions. Based on the weighted non-binary mutation matrix, a new identification model is proposed through defining new measurements of coverage and exclusivity. Then, a cooperative coevolutionary algorithm CGA-MWS is put forward for solving the presented model. Both real cancer data and simulated one were used to conduct comparisons among methods Dendrix, GA, iMCMC, MOGA, PGA-MWS and CGA-MWS. Compared with the pathways identified by the other five methods, more genes, belonging to the pathway identified by the CGA-MWS method, are enriched in a known signaling pathway in most cases. Simultaneously, the high efficiency of method CGA-MWS makes it practical in realistic applications. All of which have been verified through a number of experiments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cancer is a serious disease that has become one of the leading causes of human death. Scientific researches have demonstrated that the alterations in genome are closely related to the formation and progress of cancer [1]. All of these genome mutations can be categorized into two types, i.e., ‘driver mutations’ triggering the infinite proliferation and spread of cancer cells [2], and ‘passenger mutations’ being irrelevant to cancer [3]. Distinguishing driver mutations from passenger mutations will shine a light on understanding cancer pathogenesis and developing anti-cancer drugs [3]. The rapid development of deep sequencing technology has produced incredible amounts of multi-omics cancer data [4, 5], which turns it into reality to identify cancer related driver mutations [6]. However, many studies discovered that due to substantial mutational heterogeneity in cancer genomes, little overlap can be found between the gene mutations of two samples even though they are from the same patient [7, 8].
It is well known that there exist many significant cellular signaling or regulatory pathways in the human body, which play key roles in cell regulation including proliferation, metabolism, and apoptosis [9]. The aberration of any one driver gene involved in the pathway is usually enough to perturb its regulatory function, and lead to the formation of tumors. This may account for why the mutational heterogeneity exists. Consequently, it is essential to study mutations from the perspective of pathway level rather than gene level, which is necessary for capturing the heterogeneity in cancers [10, 11]. Three types of identification problems, such as individual driver pathway, cooperative driver pathways, and pan-cancer driver pathways, have drawn much attention. The first one is studied in this paper.
At present, there are mainly two kinds of methods for identifying individual driver pathway, i.e., prior knowledge-based methods and de novo ones. The prior knowledge-based methods try to detect a group of genes with the assistance of known biological networks. However, the incomplete and noisy of the prior knowledge may limit the detection of new combinations of mutated genes [12].
It is crucial to study the identification problem with a de novo method. Two fundamental characteristics possessed by driver pathways, i.e., high coverage and high exclusivity, are usually adopted in this kind of methods [13]. High coverage indicates a driver pathway covers a great quantity of cancer samples, whereas high exclusivity indicates each cancer sample contains just one driver gene in a pathway. Vandin et al. [12] first presented the maximum weight submatrix problem based on the two properties. Dendrix, a method based on Markov Chain Monte Carlo (MCMC), was devised to solve this problem. Zhao et al. [14] put forward a binary linear programming algorithm and a genetic algorithm (it is called as GA in this paper), both of which showed competitive performance compared with Dendrix. The GA algorithm was also easily applied to solve their presented integrative model that incorporates the gene expression profiles. Zhang et al. [15] presented a network-based approach iMCMC by integrating copy number variations (CNVs), somatic mutations, and gene expressions. Zheng et al. [16] devised a more reliable algorithm MOGA by coordinating high coverage and high mutual exclusivity. Recently, Wu et al. [17] redefined the model of the maximum weight submatrix problem, modulating coverage and mutual exclusivity by using the average weight of genes in one pathway. Then they presented a pathenogenetic algorithm PGA-MWS for solving this model.
Among the above mentioned approaches, most of which try to alleviate the negative effects from the noise in mutation data with the help of other omics data, and generate the identification model based on the two characteristics of a driver pathway. Nevertheless, it is quite usual that only mutation data is considered in the calculation of coverage and exclusivity, while the other omics data is used to calculate the weight of genes to indicate whether they are important or not. In this paper, a novel method is introduced, which measures coverage and exclusivity with not only mutation data but also gene expression data. The main contributions are depicted as follows: (1) Introduce a novel method to integrate CNVs, somatic mutations and gene expressions, and construct a weighted non-binary mutation matrix. (2) Design a new identification model by defining new measurements for coverage and exclusivity. The model may provide new ideas for identifying driver pathways from a non-binary mutation matrix. (3) Devise an identification algorithm CGA-MWS based on cooperative coevolutionary genetic algorithm. Based on the new identification model, the CGA-MWS algorithm is able to detect many gene sets having biological meaning, which has been demonstrated by extensive experiments.
2 Definitions and notations
This section begins with the integration of multi-omics data, and then an identification model is proposed. Suppose that \(S_{|P|\times |G_{S}|}\) is a somatic mutation matrix, \(C_{|P|\times |G_{C}|}\) is a CNV one, and \(E_{|P|\times |G_{E}|}\) and \(\bar {E}_{|\bar {P}|\times |G_{E}|}\) are two gene expression ones. The rows of these matrices denote a group of cancer samples P or normal samples \(\bar {P}\), and the columns of them denote a group of candidate genes GS, GC and GE, respectively. In matrix S, sij= 1 (i= 1,2,…,|P|, j= 1,2,…,|GS|) denotes that the j th gene mutates in the i th sample, and sij= 0 otherwise. Matrices C, E and \(\bar {E}\) are three real ones, where each entry of them represents relative variation or expression level of a given gene in a particular sample.
Construct a |P|×|GA| binary mutation matrix A, where GA=GS ∪ GC. Let aij= 1 when the j th gene mutates in the i th sample or it is in a statistically significant variation region of the i th sample [14], and aij= 0 otherwise (i= 1,2,…, |P|, j= 1,2,…, |GA|). The common gene set G is generated for further integrating the mutation data and the expression one, i.e., G=GA ∩ GE, and the new mutation matrix and expression ones are still presented as A|P|×|G|, E|P|×|G| and \(\bar {E}_{|\bar {P}|\times |G|}\) for convenience of description.
Given matrix E, a |P|×|G| difference matrix D=(dij) is defined. The entry dij (i= 1,2, …, |P|, j= 1,2,…, |G|) measures the difference of eij against the average expression of the j th gene among all of the normal samples, i.e., dij=\(|log_{2}e_{ij} -log_{2}\bar x_{j}|\), where \(\bar x_{j}=|\bar {P}|^{-1}\cdot {\sum }_{i=1}^{|\bar {P}|}{\bar {e}}_{ij}\). Then each entry aij in the mutation matrix A can be weighted into a float value in terms of the difference value given by dij (i= 1,2,…,|P|, j= 1,2,…,|G|), i.e., the greater aij is, the more probability of the j th gene mutates in the i th sample, as shown in (1):
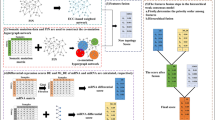
where lj=\(\max \limits \){dij|1 ≤ i ≤|P|},λ1 and λ2 are two thresholds compared with difference values (λ2 > λ1). Then the weighted non-binary mutation matrix A comes into being. aij= 1.5 means the credibility of the j th gene mutating in the i th sample is further increased, and aij=\(\frac {d_{ij}}{2\times l_{j}}\) represents that the j th gene may become a potentially important gene since its gene expression value in the i th cancer sample is much greater than the average one among normal samples. The schematic diagram for constructing the weighted non-binary mutation matrix is displayed in Fig. 1.

Schematic diagram for constructing the weighted non-binary mutation matrix. Firstly, a binary mutation matrix A is constructed by combining the somatic mutations in matrix S and CNVs in matrix C, and a difference matrix D is constructed from cancer expression data in matrix E and normal expression data in matrix \(\bar {E}\). Then a weighted non-binary mutation matrix A is obtained from the integration of matrices A and D
Assume that M is any |P|× K submatrix of A, for the i th (i= 1,2,…,|P|) row in matrix M, let η(i)=max{aij|a−j ∈ M} record the maximum weight value in the row. ν(i), a value based on Coefficient of Variation (CV), is used to measure the dispersion degree of weights in the i th row, as shown in (2):
where σ(i) and μ(i) denote the Standard Deviation and Mean Value of weights in the i th row, respectively. The greater ν(i) is, the greater dispersion degree of weights is in the i th row. Then CO(M)=\(\sum \limits _{i=1}^{|P|}\eta (i)\), ME(M)=\(\sum \limits _{i=1}^{|P|}\nu (i)\) are used to measure the coverage and mutual exclusivity of matrix M, respectively.
Based on the above definitions, a new identification model can be constructed: given a |P|×|G| weighted non-binary mutation matrix A and a parameter K (0< K < |G|), determine a |P|× K submatrix M by maximizing the weight function W (M):
3 Algorithm CGA-MWS
In this section, a cooperative genetic algorithm CGA-MWS is put forward. The input is a weighted non-binary mutation matrix A|P|×|G|, and a parameter K. The output is a |P|× K submatrix of M. We begin with describing the critical techniques in designing algorithm CGA-MWS, and then the pseudo code of it is given.
3.1 Chromosome encoding
A chromosome, namely an individual, is used to represent a solution to the problem. In the CGA-MWS algorithm, a chromosome X={x1, x2, …, xK}(xi ∈{1,2,…, |G|}, i= 1,2,…, K) is encoded by a group of K genes. It can be initialized as follows: (1) Produce a random permutation of the numbers 1 to |G|, each number denotes a gene in the matrix A. (2) Select the former K genes to construct an initial chromosome.
3.2 Fitness function
Since chromosomes correspond to feasible solutions to the identification problem, a fitness function should be defined to estimate chromosomes according to how well they perform on the problem objectives. Let MX represent the submatrix of M corresponding to chromosome X, i.e., MX is a matrix with |P| rows and |X| columns. W(MX) is used to measure the fitness of chromosome X, and Fitness(X) is defined as in (4). The greater Fitness(X) is, the better the solution X is.
3.3 Selection operator
In general, a more diversity and not deteriorate population is desired for genetic algorithm. Elitist strategy as well as roulette wheel selection are used, remaining the individual with the highest fitness in the evolution population, and picking the individual with higher fitness from parent population to participate in other evolutionary processes.
3.4 Crossover operator
Since the crossover operator can improve global search capability of genetic algorithm, a problem dependent crossover operator is proposed to inherit properties from parents to offsprings. Given a pair of parent individuals X1 and X2, fetch the common genes of them into two offspring individuals \(\hat {X}_{1}\) and \(\hat {X}_{2}\). Randomly generate a uniform order for the genes remained in X1 and X2, as well as a binary string whose length equals to half of the number of the remained genes. Each adjacent pair of genes is respectively allocated into \(\hat {X}_{1}\) and \(\hat {X}_{2}\) according to each bit of the generated binary string. For example, assume that X1={1,4,5} and X2={1,6,7}, the common gene ‘1’ is firstly fetched into \(\hat {X}_{1}\) and \(\hat {X}_{2}\), i.e., \(\hat {X}_{1}\)={1} and \(\hat {X}_{2}\)={1}. The remained genes {4,5,6,7} are shuffled randomly, and an uniform order 〈5,4,7,6〉 is determined. Let ‘10’ be the random binary string, the adjacent pair of genes 〈5,4〉 is allocated into \(\hat {X}_{1}\) and \(\hat {X}_{2}\) in terms of ‘1’, and 〈7,6〉 is allocated into \(\hat {X}_{2}\) and \(\hat {X}_{1}\) in terms of ‘0’. Then the final \(\hat {X}_{1}\)={1,5,6} and \(\hat {X}_{2}\)={1,4,7} are obtained.
3.5 Mutation operator
Although the mutation operator plays an auxiliary role in generating new individuals, it determines the local search ability in genetic algorithm. A mutation operator based on the greedy strategy is devised as follow. Given an individual X, a candidate gene set HX={g|g ∈ G, g∉X} is identified. Randomly drop a gene g from X to generate the offspring individual \(\hat {X}\), i.e., \(\hat {X}\)=X −{g}. Then the optimal gene g is extracted from any one \(\lceil \sqrt {|H_{X}|}\rceil \)-size subset \({H_{X}^{s}}\) of HX based on a greedy strategy. It means that the gene g=\(\underset {g\in {H_{X}^{s}}}{argmax} Fitness (\hat {X} \cup \){g}) is inserted into \(\hat {X}\) to obtain the final offspring individual. For example, given X={1,4,5}, HX={2,3,6,7}. The gene ‘1’ is randomly deleted from X to generate \(\hat {X}\)={4,5}. Let \({H_{X}^{s}}\)={6,2} be any one subset with size of 2, then gene 2 is inserted into \(\hat {X}\)={4,5} due to Fitness({4,5,2})(30.2) is greater than Fitness({4,5,6})(20.3).
3.6 Cooperative operators
Generally, cooperative operators can be leveraged to construct the cooperation between populations, so as to enhance the population diversity, and to avoid premature convergence and falling into local optima. In this study, two cooperative operators are devised as follows: (1) Create a cooperative pool which is composed of the individuals from all of the populations. Perform selection and crossover operators on the individuals in the cooperative pool, and produce half of the offsprings for all populations. (2) Compare the best and the worst individuals between each pair of populations, and replace the worst individual with the best one when the best one has the higher fitness. The detailed cooperative process is displayed in Algorithm 1, where Step 2, Step 5 and Step 8 describe the first cooperative operator, and Step 10, Step 11, Step 12 and Step 13 describe the second one.
3.7 CGA-MWS
In Algorithm 1, the CGA-MWS algorithm is summarized. In step 1, some parameters used in algorithm CGA-MWS are set. In step 2, the initial populations and cooperative pool are generated. Step 3 to Step 16 execute the cooperative evolution. In Step 4, selection and crossover operators are performed on parent populations to generate half of the offsprings for the new populations, respectively. Step 5 performs cooperation to generate the other half of offsprings for the new populations by using the first cooperative operator. After mutation operator is performed in Step 6, the new populations and cooperative pool are constructed in Step 7 and 8. Step 10 to Step 13 make pop1 cooperate with pop2 by using the second cooperative operator. Finally, the best individual in current generation is recorded if it is better than the best individual of the whole evolution. The whole evolution is controlled with two parameters, i.e., the maximum evolution generation maxg and the threshold of generation maintaining unchanged optimal solution maxt.

4 Results
In the experiments, real biological datasets as well as simulated ones were leveraged to compare the identification performance of the Dendrix [12], the GA [14], the iMCMC [15], the MOGA [16], the PGA-MWS [17] and the CGA-MWS methods. All of the experiments were performed on a Lenovo PC with Intel(R) Core(TM) i5-6500 3.20GHz CPU and 16GB RAM. The operating system was Windows 10, and the compiler was JAVA 8 in MyEclipse 2016 CI.
4.1 Biological data
Both glioblastoma (GBM) and ovarian datasets were adopted, where the mutation data were obtained from Zhao et al. [14], and the expression data were obtained from TCGA (http://tcga-data.nci.nih.gov/tcga/). The parameters of the CGA-MWS method were set as follows: N=\(\frac {|G|}{4}\), maxg= 1000, maxt= 10, Pm= 0.3, λ1= 3 and λ2= 7, which were determined from a large number of experiments. The parameters of methods Dendrix, GA and PGA-MWS were set as in the literatures. The Dendrix algorithm was run for 106 iterations and sampled a set every 103 iterations. The parameters of algorithm GA were set as: maxg= 1000, maxt= 10, N=|G|, pm= 0.1, and those of algorithm PGA-MWS were set as: maxg= 500, maxt= 10, N=\(log_{2}(\prod \limits _{i=0}^{K-1}|G|\)-i). The gene sets identified by algorithms iMCMC and MOGA were respectively obtained from [15] and [16], for we have not acquired the source code of them. The significance of identification results were evaluated with a random test [15]. Let W (M) be the weight score for the submatrix M with K identified genes, and W(Mi) be that for the submatrix Mi with K randomly selected genes. The process of random selection was repeated for 1000 times. The significance of submatrix M is calculated as (5).
4.1.1 Glioblastoma
In the GBM dataset, the number of samples for copy number variation, somatic mutation, normal gene expression and cancer gene expression was 206, 91, 10 and 529, respectively. After processing these data, 90 cancer samples, 10 normal samples, and 1126 genes were retained. Moreover, the genes mutating less than 1% in the samples were removed, and 920 genes were remained. In Table 1, the identified gene sets are displayed, where K ranges from 2 to 6. The genes enriched in the same biological pathway are indicated in bold, which is the same in the following tables. W denotes the score W (M) of detected gene set M.
When K was set to 2, as described by Zhao et al. [14], method GA began with identifying two optimal gene sets from the binary mutation matrix. One was (CDKN2A, TP53), and the other was CDKN2B and a metagene including CDK4 as well as TSPAN31. Then gene CDK4 was selected, for CDKN2B had stronger relationship with CDK4 than with TSPAN31, which was calculated with gene expression data. It has really been reported in KEGG database that gene set (CDKN2B, CDK4) is the part of the RB signaling pathway (Figure 2), while the evidence supporting for the association of TSPAN31 and CDKN2B is not clear. The presented method CGA-MWS is able to detect more significant gene set (CDKN2B, CDK4) (W (⋅)= 148.6) while exclude (CDKN2B, TSPAN31) (W (⋅)= 147.6) directly, even though CDK4 and TSPAN31 mutate in the same samples. Each identified gene set, except for (CDKN2A, CYP27B1) detected by the iMCMC method, is enriched in a biological pathway.

Biological pathways involved with the gene sets identified by CGA-MWS (GBM dataset). The real line indicates genes interact directly according to the KEGG database, and the dash one otherwise. The pink nodes represent the gene sets identified by method CGA-MWS. They have the same meaning in the following figures
When K= 3, gene set (CDKN2B, CDK4, RB1), part of the RB signaling pathway (Fig. 2), is identified by method GA and Dendrix. Methods PGA-MWS and CGA-MWS identify the same set (CDKN2A, CDK4, TP53), which is recorded in KEGG as the part of p53 signaling pathway (Fig. 2). It has been regarded that the mutation of gene TP53 reduces tumor suppressor activity and promotes the growth of tumors [18], and GBM cell lines having inactivated mutant p53 are more resistant to DNA-damaging therapeutic drugs [19]. In the gene set identified by methods iMCMC or MOGA, there are just two genes involving in the same biological pathway.
As implemented by [14], the genes removed by them were also dropped before performing the tests with K= 4 to K= 6, so as to identify additional gene set. Since method MOGA detects set (CDKN2B, RB1, CDK4, ERBB2) without dropping these genes, its result is omitted here. The gene sets identified by methods GA, PGA-MWS and CGA-MWS are all enriched in the PI3K-Akt signaling pathway (Fig. 2), which plays a significant role in regulating cell proliferation and is regarded as a potential target for preventing and treating metastatic tumors [20]. In contrast, in the gene set (PTEN, PIK3R1, PIK3CA, SH3YL1) detected by the Dendrix method, the first three genes are enriched in the PI3K-Akt signaling pathway, and in the gene set (EGFR, MDM2, NF1, CHAT) detected by the iMCMC method, only two genes (EGFR, MDM2) are enriched in the PI3K-Akt signaling pathway. For K= 5 and K= 6, neither iMCMC nor MOGA has reported the identification result in the literatures, hence the results of them are also omitted here. From Table 1 we can see that when K= 5 and K= 6, the gene sets identified by the CGA-MWS algorithm are all in the PI3K-Akt signaling pathway (Fig. 2). The gene PDGFRA is a direct and functionally consequential target of miR-34a, which has specifical effect on the growth of proneural glioma cells [21]. The gene COL1A2 is a critical gene in the local adhesion pathway, it may be a potential target gene for the diagnosis of GBM [22]. By contrast, in the gene sets identified by methods GA, Dendrix and PGA-MWS, there are still only a part of genes involving in the same biological pathway.
Significant tests were implemented for the five gene sets detected by method CGA-MWS. Since their p-values are less than 0.001, they are all statistically significant. Figure 3 demonstrates coverage as well as mutual exclusivity of these detected sets, where the white bars denote no mutation, the red ones denote mutually exclusive mutations, and the light green ones denote co-occurring mutations.

Results for the GBM dataset
4.1.2 Ovarian carcinoma
In the ovarian carcinoma dataset, the number of samples for copy number variation, somatic mutation, normal gene expression and cancer gene expression was 489, 316, 8 and 568, respectively. After processing these data, 313 cancer samples, 8 normal samples, and 5385 genes were retained. Similarly, the genes with mutation frequencies lower than 1% in the sample were dropped out, and 1416 genes were kept in the three matrices. Genes TP53 and TTN were filtered out from the dataset, for TP53 mutations are very prevalent (mutating in more than 80% of the samples), and TTN mutations are probably to be artifacts [14]. Table 2 shows the identification results with different settings for parameter K. The results of method MOGA are omitted for Zheng et al. [16] have not offer the detected sets.
When K= 2, methods GA, Dendrix, PGA-MWS and CGA-MWS identify the same gene set (MYC, CCNE1) covering 127 samples. Gene MYC is a strong proto-oncogene, which is usually continuously expressed in many cancers [23]. The amplification of gene CCNE1 relates to poor survival [24], which indicates it is able to become a potential therapeutic target for treating ovarian carcinoma. Both of these genes are enriched in PI3K-Akt signaling pathway (Fig. 4). The two genes (KRAS, PPP2R2A) detected by method iMCMC are not enriched in the same biological pathway.

Biological pathways involved with the gene sets identified by CGA-MWS (ovarian carcinoma dataset)
Methods GA, Dendrix, PGA-MWS and CGA-MWS initially still identified the same gene set (MYC, CCNE1, NINJ2) with K= 3. Since gene NINJ2 was not enriched in the same biological pathway along with the former two genes, we dropped it and applied these four methods again with K= 3. The new identification gene sets are displayed in Table 2. It is discovered that the genes in set (MYC, CCNE1, KRAS), detected by method CGA-MWS, are all enriched in PI3K-Akt signaling pathway (Fig. 4). Nevertheless, in respect of the other four methods, only two genes of the identified sets involve in the same pathway.
Prior to implementing the tests with K= 4 to K= 6, the genes removed by Zhao et al. [14] were also dropped. The results of method iMCMC are omitted here, for they have not been reported in [15]. As performed by Zhao et al. [14], when K= 4, method GA was applied with parameter λ= 1, and gene set (RYR2, PPP2R2A, KRAS, PRPF6) was obtained. Then it was further implemented with λ= 10 for detecting the gene set having stronger correlations, and identified (KRAS, MAPK8IP2, NF1, STMN3), as shown in Table 2. Methods Dendrix, PGA-MWS and CGA-MWS initially identified the same gene set (RYR2, PPP2R2A, KRAS, BRD4), where genes RYR2 and PPP2R2A are two significant genes engaged in the adrenergic signaling pathway (Fig. 4), and gene BRD4 is considered as a potential therapeutic target for ovarian carcinoma [25]. We removed the genes (RYR2, PPP2R2A, BRD4), belonging to the difference set between (RYR2, PPP2R2A, KRAS, BRD4) identified by Dendrix, PGA-MWS and CGA-MWS and (KRAS, MAPK8IP2, NF1, STMN3) detected by GA, and apply methods Dendrix, PGA-MWS and CGA-MWS to obtain the additional gene sets (KRAS, MAPK8IP2, NF1, STMN3), (KRAS, MAPK8IP2, NOTCH3, PRPF6) and (KRAS, MAPK8IP2, NOTCH3, MUC16), respectively. It is well known that genes KRAS, NOTCH3 and MAPK8IP2 are enriched in Apelin signaling pathway (Fig. 4), which may promote the angiogenesis and development of ovarian cancer, and be a potential pathway to target ovarian TME [26]. Gene MUC16 has been demonstrated to regulate the growth, tumorigenesis and metastasis of epithelial ovarian carcinoma cells [27].
The genes detected with either K= 5 or K= 6 can not engage in a certain pathways together. As shown in Table 2, when K= 6, among the gene set identified by the CGA-MWS method, the former four genes KRAS, MAPK8IP2, NOTCH3 and NF1 involve in Apelin signaling pathway (Fig. 4), and gene PRPF6 has been regarded as presumably critical for cancer cell survival and proliferation [28]. Since the p-values of the five gene sets detected by method CGA-MWS are less than 0.001, they are all statistically significant. Figure 5 exhibits the coverage and mutual exclusivity of these detected sets under different K values.

Results for the ovarian carcinoma dataset
4.2 Simulated data
In this section, experiments on simulated data were carried out to compare the execution efficiency of these methods for solving large size data sets. As described by Zhao et al. [14], a simulated binary mutation matrix, covering |P| patient samples and |G| genes, was generated. A group of submatrices M1, M2, …, MI (I ≥ 1) was chosen randomly, where each one contains r genes. For each sample, a gene was selected randomly from Mi (i= 1, 2, …, I) to mutate with rate pi= 1-i ⋅Δ. When a gene in matrix Mi was mutated, the other genes of this matrix mutated with rate p0. p0 and pi respectively regulate the exclusivity and coverage of Mi. The genes that were not contained in any submatrix mutated in at most three randomly selected samples. The data complexity can be adjusted by parameter I, and the increase of I increase the complexity of both the simulated data and the problem. In this study, r= 10, p0= 0.04, and Δ= 0.05, which are the same as [14]. |P|∈{100, 200, …, 1000}, |G|∈{1000, 2000, …, 10000}, I∈{1, 5, 10}. Furthermore, based on the binary mutation matrix, a non-binary mutation matrix was constructed as follows: each entry in the binary mutation matrix was changed to a non-binary one with a probability of 0.1. If the binary entry is 0, it was changed to a value of uniform distribution between 0 and 0.5, otherwise it was changed to 1.5. For each parameter setting, ten data sets were generated, and the average over ten runs at each parameter setting was calculated and presented.
We began with just comparing the execution efficiency of algorithms without considering the identification models, i.e., the running time was compared among the Dendrix algorithm [12], the GA algorithm [14], the MOGA [16], the PGA algorithm [17] and the presented CGA algorithm when they were used to solve the model of the maximum weighted submatrix problem [12]. Then the efficiency of method CGA-MWS was also tested, i.e., the execution time of the CGA algorithm on dealing with the presented model. Since the iMCMC algorithm is not related with the size of gene set K, it was not put into the comparison here. As mentioned above, the source code of algorithm MOGA were not obtained, hence it was implemented with JAVA language.
It is accepted that the execution efficiency is closely related to algorithm’s iteration times, which is affected by the convergence of the algorithm. In Figure 6, the relationship between the fitness and the number of iterations are illustrated for five genetic algorithm based methods, where I= 10, K= 10. From this figure we can see that all of the five algorithms present good convergence. The complexity of problems may increase the number of iterations required for convergence, hence the iteration times of algorithms CGA and CGA-MWS were set to 1000 for tackling problems with different complexity. In addition, the threshold of generation maintaining unchanged best solution was used to avoid useless iteration, i.e., the iteration exits when the best solution remain unchanged for a certain iteration times. In the following experiments, all of the parameters of these algorithms were set as described in the experiments on biological data. Since algorithm MOGA is devised based on the GA algorithm, it had the same parameter setting as the GA algorithm.

the relationship between the fitness and the number of iterations (I= 10, K= 10). a the GA algorithm. b the MOGA algorithm. c the PGA algorithm. d the CGA algorithm. e the CGA-MWS algorithm
In Fig. 7, the execution time under different model complexity and gene number (I= 1, 5, 10) is compared, where |P|= 500 and K= 2. From this figure, we can easily see the Dendrix algorithm runs the slowest among the five algorithms. The execution time of algorithms GA, MOGA, PGA and CGA all increases apparently with the increase number of genes, and the execution time of algorithms GA and MOGA increases faster than that of algorithms PGA and CGA. It is also noticed that the execution efficiency is not decreased with the increase of problem complexity. For example, in Fig. 7a-c, the execution time of algorithms Dendrix, GA, MOGA, PGA and CGA ranges from 422.9s to 629.3s, 0.4s to 41s, 1.2s to 111.1s, 0.6s to 14s, and 0.12s to 10s, respectively. In addition, under different combinations of I and |G|, the CGA algorithm exhibits the highest execution efficiency among the three algorithms.

Efficiency comparisons with different |G| and I. a I = 1. b I = 5. c I = 10
Figure 8 shows how the execution time scales with the increase number of patient samples |P| under different I and |G|, where K= 2. It is found that the execution efficiency is still effected by parameter I slightly. With the increase of |P|, the execution time of algorithm PGA and Dendrix increases obviously, while that of the other three algorithms increases slightly or fluctuates between a narrow range. It is also discovered that when |G|= 1000, the CGA algorithm spends the least amount of time under different |P| and I. However, when |G|= 5000 and |G|= 10000, the PGA algorithm performs the best in case of small size of samples, while the CGA algorithm does in case of large size of samples.

Efficiency comparisons with the increase of |P| under different |G|, I. a |G| = 1000 cv , I = 1. b |G| = 5000, I = 1. c |G| = 10000, I = 1. d |G| = 1000, I = 5. e |G| = 5000, I = 5. f |G| = 10000, I = 5. g |G| = 1000, I = 10. h |G| = 5000, I = 10. i |G| = 10000, I = 10
In Fig. 9, nine groups of parameters were set in dealing with parameter K, where |G|= 10000, |P|= 1000 and I= 10. As shown in this figure, with the increase of K, the execution time of algorithm PGA increases sharply, while that of algorithms Dendrix, GA, MOGA, CGA and CGA-MWS has a slight increase. Among the six algorithms, the CGA one presents the best execution efficiency, and the running time is only 58.85s even when K= 10. The CGA-MWS algorithm, applying algorithm CGA on the presented model, also exhibits high execution efficiency under different K, hence it is feasible for realistic applications.

Efficiency comparisons with different K
5 Discussion and conclusion
The identification of mutated driver pathways has received widely concerned in bioinformatics due to its importance for cancer related studies. In this paper, three types of omics data are applied to the identification. The information harboured in gene expression data is fused into the mutation data by a novel means of weighting each entry in the mutation matrix. Based on the new measurements of coverage and mutual exclusivity for the non-binary mutation matrix, a new identification model is devised. Then a cooperative coevolutionary algorithm CGA-MWS is presented for solving this model. The proposed method of integrating multi-omics data as well as the new definitions of of coverage and exclusivity for the non-binary mutation matrix may offer new ideas for other related studies.
Abundant experimental comparisons were implemented by using both biological data of glioblastoma and ovarian cancer, and simulated one. In comparison with methods Dendrix, GA, MOGA, iMCMC, PGA-MWS, the pathway identified by the CGA-MWS method usually contains more genes engaging in a known signaling pathway. In addition, the comparison results on simulation data indicate that method CGA-MWS still performs with high efficiency even when solving large-scale data, making it feasible in realistic applications. It may become a beneficial complement tool for identifying cancer pathways.
References
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70. https://doi.org/10.1016/S0092-8674(00)81683-9
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3:453–458. https://doi.org/10.1038/nrc1098
Greenman C, Stephens P, Smith R et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446:153–158. https://doi.org/10.1038/nature05610
McLendon R, Friedman A, Bigner D et al (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. https://doi.org/10.1038/nature07385
Hudson T, Anderson W, Aretz A et al (2010) International network of cancer genome projects. Nature 464:993–998. https://doi.org/10.1038/nature08987
Dees ND, Zhang QY et al (2012) Music: identifying mutational significance in cancer genomes. Genome Res 22:1589–1598. https://doi.org/10.1101/gr.134635.111
Ding L, Getz G, Wheeler D et al (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455:1069–1075. https://doi.org/10.1038/nature07423
Lawrence M, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218. https://doi.org/10.1038/nature12213
Hahn WC, Weinberg RA (2002) Modelling the molecular circuitry of cancer. Nat Rev Cancer 2:331–341. https://doi.org/10.1038/nrc795
Boca SM, Kinzler KW, Velculescu VE et al (2010) Patient-oriented gene set analysis for cancer mutation data. Genome Biol 11:1–10. https://doi.org/10.1186/gb-2010-11-11-r112
Efroni S, Ben-Hamo R, Edmonson M, Greenblum S, Schaefer CF, Buetow KH (2011) Detecting cancer gene networks characterized by recurrent genomic alterations in a population. PLoS ONE e14437:6. https://doi.org/10.1371/journal.pone.0014437
Vandin F, Upfal E, Raphael BJ (2012) De novo discovery of mutated driver pathways in cancer. Genome Res 22:375–385. https://doi.org/10.1101/gr.120477.111
Yeang CH, McCormick F, Levine A (2008) Combinatorial patterns of somatic gene mutations in cancer. FASEB J 22:2605–2622. https://doi.org/10.1096/fj.08-108985
Zhao JF, Zhang SH, Wu LY, Zhang XS (2012) Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics 28:2940–2947. https://doi.org/10.1093/bioinformatics/bts564
Zhang J, Zhang S, Wang Y et al (2013) Identification of mutated core cancer modules by integrating somatic mutation, copy number variation, and gene expression data. BMC Syst Biol 7:S4. https://doi.org/10.1186/1752-0509-7-S2-S4
Zheng CH, Yang W, Chong YW, Xia JF (2016) Identification of mutated driver pathways in cancer using a multi-objective optimization model. Comput Biol Med 72:22–29. https://doi.org/10.1016/j.compbiomed.2016.03.002
Wu JL, Cai QR, Wang JY, Liao YX (2019) Identifying mutated driver pathways in cancer by integrating multi-omics data. Comput Biol Chem 80:159–167. https://doi.org/10.1016/j.compbiolchem.2019.03.019
Warren RS, Atreya CE, Niedzwiecki D, Weinberg VK et al (2013) Association of TP53 mutational status and gender with survival after adjuvant treatment for stage III colon cancer: results of CALGB 89803. Clin Cancer Res 19:5777–5787. https://doi.org/10.1158/1078-0432.CCR-13-0351
Zhang Y, Dube C, Gibert M, Cruickshanks N, Wang B, Coughlan M, Yang Y, Setiady I, Deveau C, Saoud K, Grello C, Oxford M, Yuan F, Abounader R (2018) The p53 pathway in glioblastoma. Cancers 10:297. https://doi.org/10.3390/cancers10090297
Xu W, Yang Z, Lu N (2015) A new role for the PI3k/akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhesion Migration 9:317–324. https://doi.org/10.1080/19336918.2015.1016686
Silber J, Jacobsen A, Ozawa T, Harinath G, Pedraza A et al (2012) miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PloS one 7:e33844. https://doi.org/10.1371/journal.pone.0033844
Liang C, Zhang X, Long H, Liang CF, Fang LX, Wang G, Qi ST, Huo HZ, Song Y (2017) Prediction and analysis of key genes in glioblastoma based on bioinformatics. BioMed Res Int. https://doi.org/10.1371/journal.pone.0033844
Engler DA, Gupta S, Growdon WB et al (2012) Genome wide DNA copy number analysis of serous type ovarian carcinomas identifies genetic markers predictive of clinical outcome. PloS one e30996:7. https://doi.org/10.1371/journal.pone.0030996
Nakayama N, Nakayama K et al (2010) Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer: Interdiscip Int Jo Amer Cancer Soc 116:2621–2634. https://doi.org/10.1002/cncr.24987
Baratta MG, Schinzel AC, Zwang Y et al (2015) An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci 112:232–237. https://doi.org/10.1073/pnas.1422165112
Devapatla B K, Jaiprasart P, Dogra S, Ha J, Woo S (2016) Apelin/Apj pathway for targeting ovarian tumor microenvironment. AACR. https://doi.org/10.1158/1538-7445.AM2016-1272
Thériault C, Pinard M, Comamala M et al (2011) MUC16 (CA125) Regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecol Oncol 121:434–443. https://doi.org/10.1016/j.ygyno.2011.02.020
Chen Y, McGee J, Chen XM, Doman TN et al (2014) Identification of druggable cancer driver genes amplified across TCGA datasets. PloS one e98293:9. https://doi.org/10.1371/journal.pone.0098293
Acknowledgements
The authors are grateful to Prof. Junfei Zhao for his kindly offering the source codes of algorithm GA and the biological datasets. This research is supported by the National Natural Science Foundation of China under Grant No. 61762015, No. 61763003, Innovation Project of Guangxi Graduate Education under No. XYCSZ2020068, Guangxi Natural Science Foundation under Grant No. 2017GXNSFAA198191, Guangxi Collaborative Innovation Center of Multi-source Information Integration and Intelligent Processing, “Bagui Scholar” Project Special Funds, Guangxi Science Base and Talent Special Support No. AD16380008.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, J., Zhu, K., Li, G. et al. A model and algorithm for identifying driver pathways based on weighted non-binary mutation matrix. Appl Intell 52, 127–140 (2022). https://doi.org/10.1007/s10489-021-02330-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10489-021-02330-5