
Abstract
Ralstonia solanacearum biovar 2, a key bacterial pathogen of potato, has recently established in temperate climate waters. On the basis of isolates obtained from diseased (potato) plants, its genome has been assumed to be virtually clonal, but information on environmental isolates has been lacking. Based on differences in pulsed-field gel electrophoresis patterns, we compared the genomes of two biovar 2 strains with different life histories. Thus, genomic DNA of the novel environmental strain KZR-5 (The Netherlands) was compared to that of reference potato strain 715 (Bangladesh) by suppressive subtractive hybridization. Various strain-specific sequences were found, all being homologous to those found in the genome of reference potato strain 1609. Approximately 20% of these were related to genes involved in recombinational processes. We found a deletion of a 17.6-Kb region, denoted as a putative genomic island PGI-1, in environmental strain KZR-5. The deleted region was, at both extremes, flanked by a composite of two insertion sequence (IS) elements, identified as ISRso2 and ISRso3. The PGI-1 region contained open reading frames that putatively encoded a (p)ppGpp synthetase, a transporter protein, a transcriptional regulator, a cellobiohydrolase, a site-specific integrase/recombinase, a phage-related protein and seven hypothetical proteins. As yet, no phenotype could be assigned to the loss of PGI-1. The ecological behavior of strain KZR-5 was compared to that of reference strain 715. Strain KZR-5 showed enhanced tolerance to 4°C as compared to the reference strain, but was not affected in its virulence on tomato.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The species Ralstonia solanacearum encompasses a wide range of organisms that interact with plants, often causing plant disease. In fact, R. solanacearum has been suggested to form a true species complex (Fegan and Prior 2005), containing several types that consistently differ in biochemical properties (defining biovars), genome content and plant host range (defining races). The traditional robust classification of the organism into biovars has recently been challenged (Fegan and Prior 2005), resulting in a classification into four so-called phylotypes, denoted I through IV. Adhering to the classical taxonomy, we here will use the biovar designation.
Ralstonia solanacearum biovar 2 is a quite homogeneous group of phytopathogens (van der Wolf et al. 1998; Castillo and Greenberg 2007; Stevens and Van Elsas 2010) that cause bacterial wilt in potato as well as other crops (Janse 1998; Janse et al. 2004; Gabriel et al. 2006). It probably originated and evolved in South America, being frequently found in varying regions in Peru (Janse 1996), the cradle of potato (Ciampi and Sequeira 1980; Swanepoel 1990). In the late eighties to early nineties, the bacterium may have spread, from infested potato material, into temperate climate regions such as in the Netherlands. More than 20 years after its presumed introduction, the bacterium can still be found in Dutch local waterways, that is, in surface water, ditch sediment as well as in Solanum dulcamara (bittersweet). Microcosm experiments have shown that R. solanacearum biovar 2 can survive for relatively long periods in bulk and rhizosphere soils (Granada and Sequira 1983; Van Elsas et al. 2000), in agricultural drainage or run-off water and in canal sediment (Van Elsas et al. 2001). Furthermore, the bittersweet plants growing at the sides of waterways may serve as refuges for the organism. In these environments, the organism may show physiological responses leading to reductions in cell size, entry of (part of) the population into the viable-but-non-culturable (VBNC) state and the formation of filamentous cells and cell aggregation (Grey and Steck 2001; Álvarez et al. 2008).
During winter, R. solanacearum is often difficult to detect (as CFUs) in Dutch waterways, with only few or no typical R. solanacearum colonies appearing on semi-selective (SMSA) agar plates (Elphinstone et al. 1996). This suggests a decline in local population sizes, the emergence of VBNC cells (which are undetectable via plating), or both. When the water temperature rises again in spring and summer, bacterial cells may be released into the surface water from ditch sediment and bittersweet plants. Re-growth of the population (or resuscitation from a VBNC state) may then lead to elevated cell densities. As many crop production fields in The Netherlands are localized close to infested waterways, the biovar 2 cells present in these waters pose a continuous threat to potato production (Janse 1998; Elphinstone et al. 1998).
To understand the short-term evolution of the environmental biovar 2 populations in local waterways, genomic comparisons of recent environmental isolates with potato-derived strains are necessary. In support, the draft genomes of two plant-derived biovar 2 strains, i.e. potato strain 1609 (IPO1609; NW_002196568) and geranium strain UW551 (NZ_AAKL00000000) have very recently become available. Moreover, genomic information of the banana-derived biovar 1 strain Molk2 (YP_002254716) is also available and so is the genome sequence of biovar 3 strain GMI1000 (Salanoubat et al. 2002).
Guidot et al. (2007) investigated the R. solanacearum species complex by comparing the genome contents of 17 strains (representing all four groups of the species complex) using microarray technology. Fifty-three percent of the genes present in biovar 3 strain GMI1000 (used as the reference strain) were also present in the strains of all groups, thus defining the core gene content of the species. Very recently, it was found that variable genes are often located in mobile genetic elements, which could be characterized by lower G+C contents, or in regions of alternative codon usage, ACURs (Guidot et al. 2009b). The variable genes may have been acquired through horizontal gene transfer (HGT), while the localization of these genes is likely to be influenced by recombination events (Bertolla et al. 1999; Terol et al. 2006; Guidot et al. 2009a). Thus far, little variation at the nucleotide level has been found among the two plant-derived biovar 2 (i.e. 1609 and UW551) genomes (Gabriel et al. 2006), which might corroborate the presumed recent spread of biovar 2 from a single source. Alternatively, strong selection for a particular genetic make-up may have been required for efficient plant invasion (with the plant acting as the ecological/evolutionary bottleneck). However, as these genome data are based on just two strains, we ignore the true extent of genome diversity in biovar 2, in particular with respect to environmental strains. In many bacteria, genome diversification is mediated by HGT events as well as the activity of insertion sequence (IS) elements and transposons. Moreover, R. solanacearum is naturally transformable and its genome (both biovar 2 and biovar 3) contains a plethora of IS elements (Salanoubat et al. 2002). Together with recombinational hot spot (Rhs) elements (Wang et al. 1998), the IS elements have the potential to induce major chromosomal rearrangements, such as deletions, inversions, duplications and transpositions.
In a previous study (Stevens and Van Elsas 2010), we described a suite of 42 novel environmental R. solanacearum biovar 2 strains isolated from Dutch canal waters, sediment and bittersweet. Using genomic fingerprinting techniques, we compared these environmental strains with the potato-derived reference biovar 2 strains 715 (Bangladesh) and 1609 (Netherlands), and revealed a distribution among four major groups based on genetic techniques. Several new environmental strains, in particular strain KZR-5, stood out on the basis of the divergent pulsed-field gel electrophoresis (PFGE) patterns of the genomic DNA.
To better understand the putative genetic changes incurred in environmental strain KZR-5, we here decided to analyze its genome via suppressive subtractive hybridization (SSH) using tropical potato strain 715 as the comparator. We identified a set of strain-specific sequences that together revealed the excision of a genomic region in strain KZR-5. To delineate the ecological characteristics of KZR-5 in comparison to the reference strain, we assessed its virulence on tomato and its population dynamics in water at two ecologically-relevant temperatures.
Materials and methods
Bacterial strains
The R. solanacearum strains used in this study are listed in Table 1. Bacterial strains were stored in 20% glycerol at −80°C. Prior to each experiment, cultures from this stock were grown in 0.1× TSBS (10% strength Trypticase Soy Broth (Becton Dickinson and Company, MD, USA), 0.1% sucrose; pH 7.2) at 27°C with shaking at 180 rpm.
Genomic DNA isolation
Genomic DNA of strains KZR-5, 715, 1609, PA1, PA5, SA31 and SB63 was isolated using the Ultraclean™ microbial DNA extraction kit according to the manufacturer’s protocol (MoBIO Laboratories Inc. Carlsbad, UK). This yielded 50–100 ng/μl DNA of high quality, as measured via agarose gel electrophoresis followed by staining with ethidium bromide.
Suppressive substractive hybridization (SSH)
Genes unique to either R. solanacearum strain KZR-5 or 715 were identified by SSH using strain 715 as the tester and strain KZR-5 as the driver, as well as the reverse, i.e. strain KZR-5 as the tester and strain 715 as the driver. PCR SSH was performed using the CLONTECH PCR-Select bacterial genome subtraction kit (BD Biosciences, Palo Alto, CA) according to the manufacturer’s instructions with a few modifications. We performed the recommended control steps (i.e. adapter ligation) including a control subtraction using E. coli genomic DNA that was enriched with φX174/HaeIII digest (supplied in the kit). To check the efficiency of adapter ligation, we used primers PglA-F and R (Table 3) to amplify a 875 bp fragment of the polygalacturonase gene of R. solanacearum biovar 2 (RSIPO_03945), not containing RsaI sites. PCR was carried out in 25 μl mixtures containing 200 μM of each nucleotide, 2.5 mM MgCl2, 2% DMSO, 0.4 μM primer, 1 μl of template (prepared as instructed in the manual) and 200 U/ml of Taq polymerase (Roche Applied Science, Basel, Switzerland). The amplification conditions were 72°C for 2 min, 94°C for 30 s followed by 34 cycles of 94°C for 10 s, 58°C for 30 s and 68°C for 1 min with a final extension at 72°C for 5 min.
For the primary PCR using the adapter-specific primer SSH primer1 (Table 3), PCR was carried out as described above, except that we used a “hot start”. To obtain the hot start, the Taq polymerase (Roche) was pre-treated by mixing it 1:1 with a hot start Taq antibody (Takara BIO Inc. Shiga, Japan) and incubation for 10 min at room temperature. PCR was carried out as described above, except that the cycling conditions were: 72°C for 2 min, followed by 34 cycles of 94°C for 30 s, 62°C for 30 s and 72°C for 90 s (no final extension).
For the nested PCR, primers SSH nest1 and SSH nest2 (Table 3) were used. PCR was performed as described above (including a hot start), except that the template was 1 μl of a 1:40 dilution of the PCR products obtained from the primary PCR. The amplification conditions were 15 cycles of 94°C for 30 s, 68°C for 30 s and 72°C for 90 s (no final extension).
Construction of strain-specific clone libraries
Products obtained from the nested PCR described above were cloned using the pGEM-T easy vector system according to the manufacturer’s protocol (Promega Corporation, Madison USA). For transformation, we used competent E. coli DH5α cells (Invitrogen, Groningen, The Netherlands). At first, 115 white colonies of strain KZR-5 (driver strain 715, tester strain KZR-5) and 50 white colonies of strain 715 (driver strain KZR-5, tester strain 715) were PCR-analyzed using primers SP6 and T7 (Table 3); all showed to contain inserts ranging in size between 0.1 and 1.2 Kb. All PCR products were then digested with HaeIII and the digests analyzed on agarose gel to allow grouping of the inserts, thus avoiding the sequencing of duplicates. Based on the PCR/HaeIII clustering of the inserts (thereby excluding the clones with inserts that appeared to be similar in size and restriction pattern), 67 strain KZR-5 and 28 strain 715 specific inserts were selected for sequence analysis.
DNA sequencing
For sequencing, the inserts were amplified using PCR primers SP6 and T7 (Table 3). DNA sequencing using the T7 primer was performed in Applied Biosystems 3130 or 3730XL sequencers. Inserts larger than 1 Kb were sequenced by GATC-Biotech (Konstanz, Germany).
PCR amplification
PCR primers and annealing temperatures used for different purposes in this study are listed in Table 3. For PCR amplification, we used the GeneAmp PCR System 9700 (Applied Biosystems, Foster City, USA). Standard PCR reaction mixtures contained 1× PCR buffer (1.5 mM MgCl2, 10 mM Tris and 50 mM KCl, Roche Applied Science, Basel, Switzerland), 2.5 mM MgCl2, 10% DMSO, 200 μM each deoxynucleotide, 0.2 μM of each primer and 20 U/ml Taq DNA polymerase (Roche).
For amplification of large genomic regions (3–27 Kb), we used the TaKaRa La Taq polymerase kit (Takara BIO INC., Shiga, Japan), which is suitable for amplification of large DNA regions including high G+C content DNA. For amplification, we used the 2× GC buffer I supplied in the kit and the supplied dNTP mixture (final concentration 400 μM of each nucleotide) following the manufacturer’s instructions. The cycling program was as follows: denaturing at 96°C for 45 s; 30 cycles (96°C for 20 s, 68°C for 15 min) and a final extension step at 72°C for 15 min.
Southern hybridization
For Southern blotting, 5–10 μg of PstI or BamH1-digested genomic DNA was transferred to a Hybond-N nitrocellulose membrane (Amersham Biosciences Benelux, Roosendaal, Netherlands). For preparation of the DNA probes, we excised PCR products from agarose gel and purified these using the Qiaex II gel extraction kit (Qiagen Benelux B.V, Venlo, Netherlands). For each DNA probe, up to 1 μg cleaned PCR product was labelled using the DIG DNA labelling kit (Roche Applied Science, Penzberg, Germany). Hybridization (at 48°C), washing and detection were done using the DIG DNA detection kit according to the manufacturer’s recommendations (Roche Applied Science, Penzberg, Germany).
Survival in water at 4 and 20°C
The survival of strains KZR-5 and 715 in sterile water microcosms kept at 20 and 4°C was assessed as described elsewhere (van Overbeek et al. 2004). Microcosms were inoculated at densities of approximately 5.0E+6 CFU/ml
Virulence tests
Strains KZR-5 and 715 were tested for virulence on eight 4–5 week old tomato plants (Solanum lycopersicon cv. Maribel) using inoculum densities of 105 CFU/ml and 108 CFU/ml (to detect differences in virulence). Tomato plants are often used as a model system to test the virulence of R. solanacearum biovar 2 because of their high susceptibility and fast growth. Strain introduction was performed by watering the plant substrate (50 g dry weight sterile peat soil) with 25 ml of diluted (e.g. 10,000× or 10×, in sterile demineralized water) bacterial suspension that had grown overnight in liquid 0.1× TSBS. Prior to inoculation, plants were kept without added water for 2 days and roots were slightly damaged by gently moving the plant up and down in the soil. This procedure offered the correct window for virulence testing with the plant/peat combination that was available. Plants were incubated in the greenhouse at 26°C (day, 14 h)/21°C (night, 10 h), and disease development in the plants was scored at regular intervals over time using a disease matrix ranging from 0, no wilting symptoms, to 4, all leaves wilted (Winstead and Kelman 1952).
Nucleotide sequence accession numbers
The obtained SSH sequences were deposited in the Genbank GSS data library under number GS557176 to GS557233, the sequence of clone KZR-5 covering the deletion under number GQ899141 and the sequence of the IS blocks of strains 715, 1609 and UW551 under number GU586290 to GU586294.
Results
Approach and selection of strains
In a previous study, 42 novel R. solanacearum biovar 2 isolates were obtained which, together with two reference strains, clustered into four groups denoted as genomotypes (Stevens and Van Elsas 2010). We used a subset of these strains in the current study (Table 1). To allow the analysis of any genomic changes incurred in strains from the open environment, we selected the novel environmental R. solanacearum biovar 2 strain KZR-5 for comparison of its genome to that of the tropical potato strain 715 using two-way SSH. Strain KZR-5 was selected as (i) it was a fresh environmental isolate representative of current water populations and (ii) it was genetically clearly divergent from the reference potato strains 715 and 1609 on the basis of PFGE (whereas they appeared genetically identical with respect to five genes at the nucleotide level as evidenced by oligolocus sequence typing).
Analysis of putative strain-specific sequences
After removal of sequences of poor quality as well as duplicates, a total of 58 sequences (40 presumably specific for KZR-5 and 18 for 715) remained for further analysis. Sequences were analyzed using the Megablast tool available at http://blast.ncbi.nlm.nih.gov/Blast.cgi, which is optimized for highly similar sequences. All sequences obtained had closest matches with sequences of R. solanacearum biovar 2, showing best hits with particular regions of the strain 1609 draft genome (Table 2). One strain KZR-5 specific sequence (clone 2, Table 2) was also present in the strain 715 specific clone library (clone 42, Table 2). These sequences were homologous to non-coding regions in the 1609 genome, with nine nucleotides difference. The average G+C content of all strain-specific sequences was 54% (ranging from 25.2 to 66.2%, see Table 2), which is considerably lower than the 64% average of the R. solanacearum biovar 2 genome (Gabriel et al. 2006). For 38 sequences (30 specific for KZR-5 and 8 for 715), we did not find homologues (using Megablast) in strain GMI1000, which indicates that these sequences are possibly biovar-specific or, alternatively, divergent between these strains.
Of the putative strain-specific sequences, 48% (28/58) were homologous to sequences of a variety of functional genes, 18% (11/58) to genes encoding hypothetical proteins and another 14% to putative non-coding regions or as-yet-unassigned genes (8/58). In addition, almost 20% (11/59) were homologous, or closely related, to genes known to be potentially involved in genome flexibility and recombination processes (Table 2; clones 2, 5, 7, 16, 30, 31, 32 of strain KZR-5 and clones 42, 47, 50 and 58 of strain 715). These thus potentially identified Rhs-related proteins, transposases or related sequences. Four strain 715 specific sequences (clones 53–56) were found to be similar to a single gene (RSIPO_04909), which putatively encodes a protein with a conserved RelA/SpoT domain. A fifth cloned sequence (clone 52) was localized 1 Kb upstream of this presumed relA/spoT gene (Fig. 1A). As nearly 40% of all sequenced clones of strain 715 (11/28, including duplicates) localized to the region identified by the putative relA/spoT gene, we decided to place a focus on these sequences in our further analyses.

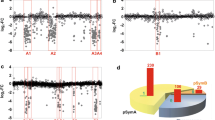
Detection of a deletion incurred in R. solanacearum biovar 2 strain KZR-5. A Localization of five sequences of strain 715 in the strain 1609 genome. The numbers 52–56 correspond to sequences of clones shown in Table 2. The spoT-F and spoT-R: indicate primers used for PCR amplification of the relA/spoT 1.6 Kb fragment. Positions 1 and 1,461 of relA/spoT (RSIPO_04909) correspond to positions 238,741 and 237,281 in the genome of strain 1609. B Southern blot analysis of genomic DNA of different R. solanacearum strains after restriction with PstI using a 1.6 Kb relA/spoT fragment as DNA probe. The R. solanacearum strains used for hybridization are indicated in the figure. Lane P is unlabeled DNA probe
Identification and characterization of a putative genomic island in R. solanacearum biovar 2 which is absent from strain KZR-5
Identification of strain-specific sequences
Using comparisons with the strain 1609 draft genome, the 11 strain 715 specific sequences were found to localize to one single genomic region of approximately 2.4 Kb in size. This region was predicted to encode two hypothetical proteins, one of which possessed a RelA/SpoT domain (Fig. 1A). To test whether the region was indeed unique for strain 715 (as well as other biovar 2 strains) and absent from strain KZR-5, we used PCR primers spoT-F and spoT-R to amplify the region from different genomes (Fig. 1A, Table 3). Using genomic DNA of strains 715, 1609, KZR-5 and four other biovar 2 strains (PA1, PA5, SA31 and SB63 (Stevens and Van Elsas 2010)), we obtained products of the expected size (1.6 Kb) for all strains except KZR-5. Southern hybridization analysis using the relA/spoT PCR fragment of strain 1609 as the DNA probe confirmed the presence of the locus in strains 715, 1609, PA1, PA5, SA31 and SB63 and its absence in strain KZR-5 (Fig. 1B).
To investigate the exact size of the deleted region in strain KZR-5, we designed eight primer sets (ps: 1–8) to amplify regions present in regions 1–20 Kb upstream as well as downstream of the relA/spoT gene (Table 3, Fig. 2A). The total region under investigation had stretches of unassigned nucleotides in the draft genome sequence of strain 1609, which indicated the presence of repeat sequences characteristic for IS elements (Fig. 2A). We used primer combinations ps9-F/ps11-R and ps6-F/ps14-R to amplify these regions, which both gave products of approximately 3 Kb in size on the basis of the genomic DNA of strains 715 and 1609. Sequence analyses of these amplicons showed that, for both strains, they consisted of IS1421 (ISRso2) and IS1021 (ISRso3) elements, with predicted transcription in opposite directions (Fig. 2B). Because we were interested in how these ISRso2 and ISRso3 elements are linked to each other in these IS “blocks”, we designed an additional primer (IS2/3-seq, Table 3) for further sequencing. For both IS blocks, we found a 107-bp sequence connecting the two IS elements, with no apparent homology to other known sequences, except for a 21-bp stretch with 100% homology to a Holiday structure resolvase of Bifidobacterium longum (bp 56–76 of the 107 bp element (Fig. 2B). Hence, although functional evidence is still lacking, the full region determined by the blocks likely contained several functions of relevance for transposition/recombination processes.

Primer systems used to characterize the identified region (A) and the composition of the IS blocks (B). A
Upper line: inferred localization of IS elements and the relA/spoT region and PCR systems based on genomic information of strain 1609. IS: unassigned nucleotides in the 1609 genome which represent insertion sequences. Superscript 1 Numbers: primer systems, corresponding to primers shown in Table 3 (ps1 through ps15; ps15 not indicated here). PCR systems consisted of two PCR primers (e.g. 1: ps1-F/ps1-R, 2: ps2-F/ps2-R, etc.). *: ps10 amplifies ISRso3. **: ps13 amplifies ISRso2s. Second line: Presence and absence of sequences in strain KZR-5 on the basis of PCR and hybridization.  : The absence in strain KZR-5 was confirmed by Southern blot analysis using the corresponding PCR products of strain 1609 as DIG labeled DNA probes. B Insertion sequence (IS) regions determined by sequence analysis of PCR products of strains 715 and 1609 that were obtained with primer combinations ps9-F/ps11-R or ps6-F/ps14-R. The position of the sequencing primers (ps9-F, ps6-F, ps11-R, ps14-R and IS2/3) is indicated. Superscript 2: IS regions correspond to the two IS regions, which are similar, shown in A (IS)
: The absence in strain KZR-5 was confirmed by Southern blot analysis using the corresponding PCR products of strain 1609 as DIG labeled DNA probes. B Insertion sequence (IS) regions determined by sequence analysis of PCR products of strains 715 and 1609 that were obtained with primer combinations ps9-F/ps11-R or ps6-F/ps14-R. The position of the sequencing primers (ps9-F, ps6-F, ps11-R, ps14-R and IS2/3) is indicated. Superscript 2: IS regions correspond to the two IS regions, which are similar, shown in A (IS)
All PCR amplifications, except those with primer sets ps5 and ps6 (Table 3), performed on genomic DNA of strains 715, 1609 and KZR-5, yielded amplicons of the expected sizes (between 150 and 600 bp). Primer sets ps5 and ps6 also yielded such products for strains 715 and 1609, but not for strain KZR-5 (Table 3, Fig. 2A), thus indicating the presence of a deletion. To more precisely determine the size of the deletion, we designed seven additional primer sets (Table 3; ps9 through ps15) for comparison of this region between the strains. Primer sets ps9, ps10 and ps13 through ps15 yielded products of the expected sizes for all tested strains, indicating that these regions were not part of the deletion. In contrast, primer sets ps11 and ps12 gave products of the expected sizes for strains 715 and 1609, whereas amplification on KZR-5 genomic DNA yielded no products (Fig. 2A).
To allow a cross-comparison of the identified genomic region between strains 715, 1609, UW551 and KZR-5, we then performed PCR with a range of different primer combinations across most of the deleted region (see Fig. 2). Using the combinations ps4-F with ps14-R/ps7-R or ps9-F with ps11-R/ps14-R/ps7-R, amplification products of the expected sizes (ranging from 1 to over 10 Kb) were obtained for strains 715, 1609 and UW551, as visualized on agarose gel (data not shown). However, these bands were lacking from the PCR reactions performed on strain KZR-5 (data not shown). PCR amplification across the IS blocks (using primers ps9-F/ps11-R and ps6-F/ps14-R) produced amplificons of approximately 3 Kb that were similar across strains 715, 1609 and UW551. This suggests that, for these strains, the PGI-1 region localizes within a similar genomic context.
To amplify the region around the putative deletion from strain KZR-5 genomic DNA, we used primers ps4-F/ps14-R and ps4-F/ps7-R, for which the amplicons were expected to range between 2 and 4 Kb. However, no such products were obtained. We also performed a PCR using primer pair ps11-F/ps14-R on strain KZR-5 genomic DNA, and, unexpectedly, obtained a clear 3.6 Kb product. To elucidate its sequence, the PCR product was cloned and partially sequenced using primers ps11-F, ps13-R, IS2/3 and ps14-R. Thus, the region to which primer ps11-F had annealed was identified as follows: 861 bp upstream of the ps4-F target site (Fig. 2A), the stretch tgtgcctacgttgaatga was present, which showed strong homology at the 3′-end with primer ps11-F (cttgctgcctcttgaatga) and thus a ps11-F “landing” site was identified. Alignment of the resulting sequence with that of the strain 1609 genomic region showed that a region of 19.8 Kb, spanning the complete region in between the IS elements (17.6 Kb) plus one ISRso2/ISRso3 block of 2.2 Kb (Fig. 3), was present in strain 1609 and, by inference, 715, but had been deleted from strain KZR-5. The analysis also showed that the region harbors four XbaI restriction sites, which explains the observed PFGE pattern (XbaI digested genomic DNA) seen for strain KZR-5 as compared to that of strains 715 and 1609 (Stevens and Van Elsas 2010).

The PGI-1 region in strains 1609 and UW551, the deletion in strain KZR-5 and the related regions in strains Molk2 and GMI1000. The average G+C content of PGI-1 is shown over 200 bp intervals (upper graph). The line indicates the average G+C % over the whole genome (UW551; 64%). The location of four XbaI sites (positions 3,614, 5,330, 8,156 and 13,293) is also indicated. Positions 1 and 20 Kb correspond to position 243,626 (1) and 225,013 (20 Kb) of strain 1609. The location of the IS elements and the PGI-1 region (in between the IS blocks) in strains 1609 and UW551 are indicated (second graph). The organization of this region is, based on PCR and Southern hybridization results, the same in strain 715. Numbers below arrows indicate ORF numbers as in Table 4. ORF 1 through 3 encode hypothetical proteins, ORF 4 encodes a RelA/SpoT domain protein, ORF 5 a site specific integrase/recombinase, ORF 6 a bacteriophage related protein, ORF 7 a drug metabolite transporter (DMT) protein, ORF 8 a transcriptional regulator protein, ORF 9 through 11 hypothetical proteins, ORF 12 a cellobiohydrolase and ORF 13 a hypothetical protein. The organization of the relevant regions in strains KZR-5, Molk2 and GMI1000 are also shown. * ORF 1, 11, 5 and 6 localize on the chromosome, ORF 12 localizes on the megaplasmid.  Alternative codon usage region (ACUR)
Alternative codon usage region (ACUR)
Characterization of the region
Based on information from the strain 1609 draft genome, the identified region has an average G+C content of 55%, which is considerably lower than the 64% average of the complete genome (Fig. 3). Comparison of the sequences of the PGI-1 region (17.6 Kb size in between the IS elements) in the genomes of strains 1609 and UW551 (position 25,398–43,007) showed complete identity between the two regions, although the number of genes that had been annotated was somewhat different (Table 4). According to the strain 1609 annotation, the region contained genes encoding (1) a protein with a RelA/SpoT domain, which is a putative (p)ppGpp synthetase (ORF4, RSIPO_04909), (2) a transporter protein of drugs or metabolites (ORF7, RSIPO_04908), (3) a transcriptional regulator (ORF8, RSIPO_03301), (4) a cellobiohydrolase, which is involved in the degradation of cellulose (ORF 12, RSIPO_03298) and eight hypothetical proteins (Table 4). In addition, according to the strain UW551 annotation (Gabriel et al. 2006), there was a (bacteriophage-related) site-specific integrase/recombinase (ssi/r) (ORF5, RRSL_02058), a bacteriophage-related hypothetical protein (ORF6, RRSL_02059) and one additional hypothetical protein (ORF13, RRSL_02066) (see Fig. 3 and Table 4). To further investigate genome flexibility, we assessed about 3 Kb of flanking sequence in the 1609 draft genome at both sides of the identified region. In the region flanking the left IS elements, we found a hypothetical protein and a hemagglutinin-related protein. At the right flank, we found genes homologous to those encoding TrbI (involved in bacterial conjugation), next to a Vgr-related protein (Vgr stands for a domain with valine/glycine repeats). In E. coli a protein with valine/glycine repeats and associated with Rhs elements (Wang et al. 1998), both potentially involved in genome flexibility, and two hypothetical proteins (data not shown).
To identify possible homologues of the cbhA and relA/spoT genes in the genomes of strains 1609 and UW551, we used Blast-N and Blast-P (available at NCBI) on the respective genome information. However, we did not find close homologues of the cbhA or relA/spoT genes such as found on PGI-1 in the biovar 2 genomes (based on sequence homology and annotation). In addition, Southern hybridization with a cbhA-specific DNA probe showed single bands with genomic DNA of strains 715 and 1609 and no hybridization signal in strain KZR-5, similar to what was seen for the putative gene with RelA/SpoT domain (Fig. 1B). The relA/spoT gene of biovar 2 showed highest homology (using Blast-P) to a similar gene from Rhizobium etli (46% identity) or Exiguobacterium sp. (40% identity), followed by a hypothetical phage-derived protein from E. coli (36% identity) and a putative relA/spoT gene from Symbiobacterium thermophilium (37% identity).
Comparison of the PGI-1 region across the sequenced R. solanacearum strains 1609, Molk2 and GMI1000 showed that several genes found in the region are indeed genetically flexible as they are flanked by IS elements and/or ACURs (Fig. 3). For instance, the PGI-1 region in strain Molk2 was partially identical to that of strain 1609, as ORFs 5 through 12 appeared to be conserved. However, the Molk2 region completely lacked ORFs 1 to 4 as well as ORF13, which also were not present elsewhere in the genome (Table 4, Fig. 3). Much like in strain 1609, the genes present were flanked by IS elements, but the nature of the IS elements differed. In contrast, the genes present in the PGI-1 regions of strains 1609, UW551 and Molk2 do not occur in a PGI-1-like island in the biovar 3 GMI1000 genome. In the latter strain, some of the genes are dispersed over the chromosome as well as the megaplasmid, while other genes are completely absent. For instance, the cbhA gene is encoded by a region on the megaplasmid, denoted Rsp0583. ORF 1 (hypothetical protein) and ORFs 5 (ssi/r) and 6 (bacteriophage-related protein) localize 62 Kb apart from each other on the chromosome, with ORFs 5 and 6 co-localizing, like in PGI-1.
To determine whether islands like PGI-1 exist in other bacterial genomes, which might indicate a recent HGT event, we used the “string” database (http://string.embl.de), which aligns multiple ORFs against the 2,483,276 proteins of 630 organisms. Using this approach, we did not find any genomic regions with a similar gene order in other bacterial strains. However, in the genomes of Polaromonas sp. JS666 and P. fluorescens pfO1, we found that the genes for the transporter protein (RSIPO_04908) and the transcriptional regulator protein (RSIPO_03301) localize adjacent to each other, similar to the situation in PGI-1.
Phenotype and ecological behavior of environmental strain KZR-5 in comparison to the tropical potato-derived strain 715
To assess whether the loss of the genes for the putative RelA/SpoT domain protein and the cellobiohydrolase (CbhA), both uniquely present on PGI-1, conferred a discernable phenotype in strain KZR-5, we performed standard cellulose degradation and growth tests on strains KZR-5 and 715. Surprisingly, no differences in phenotypic behavior between strains KZR-5 and 715 were found.
Then, to understand whether the novel environmental R. solanacearum strain KZR-5, in comparison to the potato-derived reference strain 715, had altered fitness in water under temperate climate conditions, we performed assessments of population dynamics at two temperatures in microcosms. Thus, the survival of strain KZR-5 was compared to that of strain 715 upon incubation at 4 and 20°C (control). Strain KZR-5 persisted in a fashion similar to strain 715 in water at 20°C, with CFU numbers remaining roughly stable, between log 6.6 and log 7.1 from the onset of the experiment till day 130 (data not shown). At 4°C, the two strains behaved quite differently, with strain KZR-5 clearly being the best survivor over the experimental period (Fig. 4A). At day 85, the CFU numbers of strain KZR-5 remained detectable at average levels between log 2.6 and 3.4 ml−1, whereas those of strain 715 were at or below the limit of detection (Fig. 4A). These counts were significantly different between strains KZR-5 and 715 (Fig. 4A; t test; P < 0.05). Thus, enhanced tolerance to cold stress was noted in strain KZR-5 as opposed to the comparator strain 715.

Survival of strains KZR-5 and 715 at 4°C (A) and virulence on tomato plants (B). A Culturability (the average of three replicates) of strains KZR-5 and 715 upon exposure to 4°C at t = 1 (bar 1) and t = 84 days (bar 2). - - - - - -, limit of detection (10 CFU/ml). B Average wilting of 4–5 week old tomato plants at 7, 14 and 21 days post inoculation. Plants were inoculated 25 ml of cell suspension containing 10+5 CFU/ml, incubated in the greenhouse at 26°C (day, 14 h)/21°C (night, 10 h), and disease development in the plants was scored at regular intervals over time using a disease matrix ranging from 0, no wilting symptoms, to 4, all leaves wilted. The average wilting index was determined as wilting symptoms of all plants/number of plants
Given the fact that a presumed cbhA gene was found to be present on PGI-1, we hypothesized that strain KZR-5 might have suffered a reduction in virulence on susceptible host plants as compared to the reference strain as well as, possibly, other environmental strains. Thus, virulence assays were performed on tomato using inoculum densities of 108 CFU/ml. The tests revealed complete wilting of the replicate test plants within 14 days (data not shown). Surprisingly, in none of the cases were significant differences in virulence found between strains KZR-5 and 715 (data not shown). As subtle differences might escape detection using high inoculum densities, we also performed a virulence assay using the same strains with inoculum densities of approximately 105 CFU/ml. At day 7, the wilting index of plants infested by strain KZR-5 was slightly higher (1.0) than that for strain 715 (0.6). At days 14 and 21, the wilting index was similar for both strains (2.8 vs. 2.9 at day 14 and 3.3 vs. 3.4 at day 21; P > 0.05), suggesting these were, at least under these conditions, equally virulent on tomato.
Discussion
From among a larger set of novel environmental R. solanacearum biovar 2 strains, strain KZR-5 was specifically selected for a comparison of its genomic make-up to that of reference potato strain 715 using suppressive subtractive hybridization (SSH). This comparative analysis was undertaken in order to allow insight in the putative genomic changes incurred in strains present in the open environment for up to two decades, versus a tropical potato strain. The basis of the selection was a divergent PFGE pattern of XbaI digested genomic DNA that was previously revealed in strain KZR-5, which already provided a glimpse of genome diversity incurred by a genomic rearrangement (Stevens and Van Elsas 2010).
Several studies have shown that SSH can be successful as long as the genomes that are subjected to the procedure are grossly homologous (Akopyants et al. 1998; Zhang et al. 2000; Janke et al. 2001; Parsons et al. 2003). Both R. solanacearum strains used here belong to biovar 2, which is known as a highly homogeneous group of organisms. Oligolocus sequence typing of over 2,000 nucleotides showed that the strains were 100% homologous in the six regions analyzed (Stevens and Van Elsas 2010). The SSH approach used in this study was effective in identifying genes or genomic regions that differ between the two selected R. solanacearum biovar 2 strains. All sequences found actually had homologues in the genome of strain 1609. Hence, we did not find sequences that had been newly acquired by strain KZR-5, pointing to an absence of major HGT events as drivers of short-term evolution in the water population exemplified by strain KZR-5.
A major finding was the presence, in the SSH library, of a large number of sequences that are known to be involved in genome flexibility (7/40 of KZR-5 and 4/19 of 715, see Table 2). This clearly points to a major role of genomic rearrangements in shaping the R. solanacearum biovar 2 genome under the local conditions. Moreover, the fact that we found a partial rRNA gene sequence in each library that localized to a single rrn operon (clones 10 and 46, Table 2) might indicate that such regions, together with the Rhs elements, mediate chromosomal rearrangements like those shown by Hill (1999). However, we have not further addressed this hypothesis.
The clearest evidence found in this study for the involvement of DNA rearrangements in genome diversification between the strains analyzed was the finding of a deletion of a putative genomic island, PGI-1, in strain KZR-5. In this case, strain 715 was the tester and strain KZR-5 the driver in the SSH analysis. We then used multiple PCR systems to, firstly, define the size of the deletion in strain KZR-5 and, secondly, amplify and sequence the flanking regions. We cannot explain why amplification with primer sets ps4-F/ps14-R or ps9-F/ps14-R was unsuccessful in strain KZR-5, while the ps11-F/ps14-R set amplified a specific product. One possible explanation could be that other copies of ISRso2 and ISRso3 sequences (multiple copies are present in the R. solanacearum biovar 2 genome), hamper PCR amplification by acting as a primer sink due to “random” or “aspecific” annealing of the primers.
The PGI-1 region has key features of an ecologically relevant genomic island that is potentially mobile because (i) it has a lowered average G+C content compared to the average G+C content of the strain 1609 genome (55 vs. 64%), (ii) it showed the presence of a site-specific integrase/recombinase (ssi/r) and a phage-related protein, (iii) there was a deletion of the region exactly at the IS blocks present at the island extremes, (iv) it revealed the presence of genes immediately at the right flank of the right ISRso3 element that encode TrbI (protein involved in conjugation) and a Vgr-related protein, which (in E. coli) associates with Rhs and (v) it revealed the presence of genes such as cbhA and relA/spoT that are potentially involved in an ecologically relevant phenotype. Although we could assign potential functions to six ORFs, the function of eight other putative ORFs that were identified on the genomic island remained largely unknown. These ORFs encoded hypothetical proteins that lack known conserved domains. Hence, we cannot make any firm inferences about the function of these proteins and the possible effect of their deletion.
Comparison of the PGI-1 region with similar ones in biovar 1 strain Molk2, and in biovar 3 strain GMI1000 provided support for the notion that a genetically flexible region was found that was (1) consistently present in biovar 2 strains 1609 and UW551 and, by inference, 715, and (2) partially and differentially present across the other biovar strains, whereas it was absent from strain KZR-5. This finding supports the hypothesis that the region is a genomic island, as the PGI-1 region was flanked by IS elements in strains 715, 1609 and UW551. Also, in the biovar 3 strain GMI1000 some genes of the island, i.e. cbhA and the ssr/i and phage-related genes, were found to lie inside, or close to, ACURs which have probably been acquired through HGT (Salanoubat et al. 2002).
As a cellobiohydrolase gene was found to be present in the reference potato-derived R. solanacearum biovar 2 strains 1609 and 715 (as well as in biovar 1 strain Molk2 and biovar 3 strain GMI1000), but absent from non-phytopathogenic Ralstonia species (Liu et al. 2005), it might play a role in the interaction of R. solanacearum with host plants. Moreover, biovar 2 strain UW551 and biovar 3 strain GMI1000 produce, next to the cellobiohydrolase, at least five other enzymes, i.e. a β 1,4-endoglucanase (Egl), an endopolygalacturonase (PglA), two exopolygalacturonases (PehB and PehC) and a pectin methyl esterase (Pme). Collectively, these enzymes probably assist the bacterium in the degradation of plant cell materials (Gabriel et al. 2006). It was shown that a GMI1000 mutant lacking the cbhA gene was reduced in virulence, corroborating the role of CbhA in the strain’s ability to wilt plants (Liu et al. 2005). The finding of the loss of cellobiohydrolase in strain KZR-5 and the concurrent lack of an effect on plant invasion was puzzling. One explanation might be that for biovar 2 strains the cbhA gene is less important in plant invasion than for biovar 1 strains. Alternatively, a functional homologue of CbhA might be present in strain KZR-5, although we did not find other cbhA-like genes in the draft genome sequences of strains 1609 and UW551, nor additional bands with Southern blot analysis using a cbhA DNA probe. Therefore, it is unlikely that gene duplication had occurred in strain KZR-5, which would have maintained the functionality of the gene in spite of its deletion with PGI-1. As endo- and exoglucanases have a glycosyl hydrolase family 6 (GH6) domain (http://www.cazy.org/), we inspected the annotated 1609 and UW551 genomes to see whether other glycosyl hydrolases (with a presumed cellobiohydrolase activity) exist in the biovar 2 genome. We found four such genes (RSIPO_01357, 03533, 04005 and 03946), but these belong to other functional groups (GH15, GH18 or the AlgLyase superfamily) than CbhA (and Egl) and have different roles in carbohydrate metabolism. However, other functional homologues might still exist in R. solanacearum biovar 2, as the function of many genes in the biovar 2 genome is unknown.
The presence of a gene encoding a protein with a RelA/SpoT domain in the deleted PGI-1 region was striking. RelA/SpoT proteins are conserved across the bacteria, as they are thought to function in responses to starvation or other stress, as a result of their ppGpp(p) synthetase/hydrolase activity (ppGpp is a so-called alarmone). In E. coli, the relA and spoT genes become activated as part of the stringent (stress) response upon amino acid (relA) and carbon (spoT) starvation. However, the function of RelA/SpoT in the stress response might differ between different bacterial species (Das and Bhadra 2008; Chatterji and Ojha 2001). In R. solanacearum, homologues of the E. coli RelA and SpoT proteins exist (RSIPO_01119 and RSIPO_01943 respectively), but they were never studied in detail. In many gram-positive bacteria, only a single bifunctional RelA/SpoT homologue is responsible for balancing (p)ppGpp levels in the cell (Mittenhuber 2001). However, in Bacillus subtilis and Streptococcus mutans, other functional ppGpp synthetases are also described (Lemos et al. 2007; Nanamiya et al. 2008). These proteins have a RelA/SpoT domain, but lack the other conserved motifs found in traditional RelA and SpoT proteins and they appear to represent a different class of (p)ppGpp synthetases called SAS (small alarmone synthetase) proteins (Nanamiya et al. 2008). A comparison of the amino acid sequence of the R. solanacearum RelA/SpoT domain protein with the SAS proteins of B. subtilis and Streptococcus mutans showed they are indeed similar in composition and size (data not shown), thus indicating a putative similar function. The SAS proteins do not appear to be essential in the classical stress response but function in the synthesis of alarmone under other conditions (Nanamiya et al. 2008). Why a RelA/SpoT domain protein, which might be associated with presumed ecological fitness (survival) under stress, was deleted from the genome of strain KZR-5 and whether this led to enhanced fitness, is still unclear from the current work and thus remains speculative. The enhanced survival of KZR-5 in water at low temperature, though, provides food for the contention that there may be an advantage for the possession of the deletion under such stress conditions. Our results may indicate that life in an aquatic environment in a temperate climate (characterized by fluctuating but largely low temperature and nutrient conditions, persistence in bulk water, sediment and/or bittersweet) has incited a different survival modus, possibly also altering the function of other genes involved.
IS elements clearly played a major role in the deletion event, through an interaction between the two ISRso2/ISRso3 blocks that flank the island. It seems likely that a recombination/cross-over occurred between the two 2.2 Kb large elements, which are 100% homologous to one another, thereby deleting a DNA loop of 17.6 Kb in between the ISRso2/ISRso3 elements. IS element mediated genome diversification could play an important role in the structural flexibility of R. solanacearum biovar 2, like in other bacterial species such as Burkholderia mallei (Nierman et al. 2004), Yersinia species (Darling et al. 2008) and Pseudomonas aeruginosa (Battle et al. 2009). Unfortunately, although tens of ISRso3 elements have recently been found using hybridisation (Stevens and Van Elsas 2010), we do not know the exact number and variability of diverse IS elements in the biovar 2 genome. We also ignore whether more IS blocks, such as found in PGI-1, exist in the genome, as these regions are often “missed” using shotgun genome sequencing as used for the biovar 2 strain 1609 draft genome.
One of our aims was to assess whether environmental strain KZR-5 was different in ecological behavior as compared to the tropical potato strain 715. First, upon inoculation of tomato plants at two inoculum densities, no changed ability of strain KZR-5 to cause wilting disease was observed, in spite of the deleted cbhA gene. The putative differences in virulence on tomato between strains KZR-5 and 715 may have been minor, which may relate to the reasons outlined above. On the other hand, the survival of strain KZR-5 in water at low temperature was clearly enhanced compared to that of strain 715. Thus, strain KZR-5 may have adapted to conditions prevailing in temperate climate waters in relation to the reference potato strain.
In this study, we pinpointed the activity of IS elements (and Rhs elements) as the main mechanism that facilitates genomic changes in R. solanacearum biovar 2 and thus its potential adaptation to selective pressures from the environment. Despite our increasing knowledge about the genetic content of whole bacterial genomes (Binnewies et al. 2006), we still largely rely on the examination of single strains, and their unique genetic make-up, to assess how a particular genetic context (including the presence or absence of genomic islands) correlates with strain behavior in the environment. Future work with biovar 2 strain KZR-5 will attempt to more precisely establish this correlation.
References
Akopyants NS, Fradkov A, Diatchenko L, Hill JE, Siebert PD, Lukyanov SA et al (1998) PCR-based subtractive hybridization and differences in gene content among strains of Helicobacter pylori. Proc Natl Acad Sci USA 95:13108–13113
Álvarez B, López MM, Biosca EG (2008) Survival strategies and pathogenicity of Ralstonia solanacearum phylotype II subjected to prolonged starvation in environmental water microcosms. Microbiology 154:3590–3598
Battle SE, Rello J, Hauser AR (2009) Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol Lett 290:70–78
Bertolla F, Frostegard A, Brito B, Nesme X, Simonet P (1999) During infection of its host, the plant pathogen Ralstonia solanacearum naturally develops a state of competence and exchanges genetic material. Mol Plant Microbe Interact 12:467–472
Binnewies TT, Motro Y, Hallin PF, Lund O, Dunn D, La T, Hampson DJ et al (2006) Ten years of bacterial genome sequencing: comparative-genomics-based discoveries. Funct Integr Genomics 6(3):165–185
Castillo JA, Greenberg JT (2007) Evolutionary dynamics of Ralstonia solanacearum. Appl Environ Microbiol 73:1225–1238
Chatterji D, Ojha AK (2001) Revisiting the stringent response, ppGpp and starvation signaling. Curr Opin Microbiol 4:160–165
Ciampi L, Sequeira L (1980) Influence of temperature on virulence of race 3 strains of Pseudomonas solanacearum. Am Potato J 57:307–317
Darling AE, Miklos I, Ragan MA (2008) Dynamics of genome rearrangement in bacterial populations. PLoS Gene 4:e1000128
Das B, Bhadra RK (2008) Molecular characterization of Vibrio cholerae ΔrelA ΔspoT double mutants. Arch Microbiol 189:227–238
Elphinstone JG, Hennessy J, Wilson JK, Stead DE (1996) Sensitivity of different methods for the detection of Pseudomonas solanacearum (Smith) Smith in potato tuber extracts. Bull OEPP 26:663–678
Elphinstone JG, Stanford HM, Stead DE (1998) Detection of Ralstonia solanacearum in potato tubers, Solanum dulcamara and associated irrigation water. In: Prior PH, Allen C, Elphinstone J (eds) Bacterial wilt disease: molecular and ecological aspects. Springer-Verlag, Berlin, pp 133–139
Fegan M, Prior P (2005) How complex is the Ralstonia solanacearum species complex? In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. American Phytopathological Society Press, St. Paul, pp 449–461
Gabriel DW, Allen C, Schell M, Denny TP, Greenberg JT, Duan YP et al (2006) Identification of open reading frames unique to a select agent: Ralstonia solanacearum race 3 biovar 2. Mol Plant Microbe Interact 19:69–79
Granada GA, Sequira L (1983) Survival of Pseudomonas solanacearum in soil, rhizosphere and plant roots. Can J Microbiol 29:433–440
Grey BE, Steck TR (2001) The viable but nonculturable state of Ralstonia solanacearum may be involved in long-term survival and plant infection. Appl Environ Microbiol 67:3866–3872
Guidot A, Prior P, Schoenfeld J, Carrere S, Genin S, Boucher C (2007) Genomic structure and phylogeny of the plant pathogen Ralstonia solanacearum inferred from gene distribution analysis. J Bacteriol 189:377–387
Guidot A, Coupat B, Fall S, Prior P, Bertolla F (2009a) Horizontal gene transfer between Ralstonia solanacearum strains detected by comparative genomic hybridization on microarrays. ISME J 3:549–562
Guidot A, Elbaz M, Carrere S, Siri MI, Pianzzola MJ, Prior P et al (2009b) Specific genes from the potato brown rot strains of Ralstonia solanacearum and their potential use for strain detection. Phytopathology 99:1105–1112
Hill CW (1999) Large genomic sequence repetitions in bacteria: lessons from rRNA operons and Rhs elements. Res Microbiol 150:665–674
Janke B, Dobrindt U, Hacker J, Blum-Oehler G (2001) A subtractive hybridisation analysis of genomic differences between the uropathogenic E. coli strain 536 and the E. coli K-12 strain MG1655. FEMS Microbiol Lett 199:61–66
Janse JD (1996) Potato brown rot in western Europe—history, present occurence and some remarks on possible origin, epidemiology and control strategies. Bull OEPP 26:679–695
Janse JD (1998) Experiences with potato brown rot Ralstonia solanacearum biovar 2, race 3 in the Netherlands. In: Prior PH, Allen C, Elphinstone J (eds) Bacterial wilt disease, molecular and ecological aspects. Springer-Verlag, Berlin, pp 146–152
Janse JD, van den Beld HE, Elphinstone J, Simpkins S, Tjou-Tam-Sin NNA, van Vaerenbergh J (2004) Introduction to Europe of Ralstonia solanacearum biovar 2, race 3 in Pelargonium zonale cuttings. J Plant Pathol 86:147–155
Lemos JA, Lin VK, Nascimento MM, Abranches J, Burne RA (2007) Three gene products govern (p)ppGpp production by Streptococcus mutans. Mol Microbiol 65:1568–1581
Liu H, Zhang S, Schell MA, Denny TP (2005) Pyramiding unmarked deletions in Ralstonia solanacearum shows that secreted proteins in addition to plant cell-wall-degrading enzymes contribute to virulence. Mol Plant Microbe Interact 18:1296–1305
Mittenhuber G (2001) Comparative genomics and evolution of genes encoding bacterial (p)ppGpp synthetases/hydrolases (the Rel, RelA and SpoT proteins). J Mol Microbiol Biotechnol 3:585–600
Nanamiya H, Kasai K, Nozawa A, Yun CS, Narisawa T, Murakami K et al (2008) Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol Microbiol 67:291–304
Nierman WC, DeShazer D, Kim HS, Tettelin H, Nelson KE, Feldblyum T et al (2004) Structural flexibility in the Burkholderia mallei genome. Proc Natl Acad Sci USA 101:14246–14251
Parsons YN, Banasko R, Detsika MG, Duangsonk K, Rainbow L, Hart CA (2003) Suppression-subtractive hybridisation reveals variations in gene distribution amongst the Burkholderia cepacia complex, including the presence in some strains of a genomic island containing putative polysaccharide production genes. Arch Microbiol 179:214–223
Salanoubat M, Genin S, Artiguenave F, Gouzy J, Mangenot S, Arlat M (2002) Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415:497–502
Stevens P, Van Elsas JD (2010) Genetic and phenotypic diversity of Ralstonia solanacearum biovar 2 strains obtained from Dutch waterways. Antonie van Leeuwenhoek 97(2):171–188
Swanepoel AE (1990) The effect of temperature on the development of wilting and on progeny tuber infection of potatoes inoculated with South-African strains of biovar-2 and biovar-3 of Pseudomonas-Solanacearum. Potato Res 33:287–290
Terol J, Munoz-Soriano V, Arahal DR, Buades C, Perez-Perez A, Llop P et al (2006) Random genome sequencing of Ralstonia solanacearum strain IVIA 1602 and comparative analysis with strain GMI1000. J Phytopathol 154:556–564
Timms-Wilson TM, Bryant K, Bailey MJ (2001) Strain characterization and 16S–23S probe development for differentiating geographically dispersed isolates of the phytopathogen Ralstonia solanacearum. Environ Microbiol 3:785–797
Van der Wolf JM, Bonants PJM, Smith JJ, Hagenaar M, Nijhuis E, van Beckhoven JRCM (1998) Genetic diversity of Ralstonia solancaerum Race 3 in Western Europe determined by AFLP, RC-PFGE and Rep-PCR. In: Prior PH, Allen C, Elphinstone J et al (eds) Bacterial wilt disease: molecular and ecological aspects. Springer-Verlag, Berlin, pp 44–49
Van Elsas JD, Kastelein P, van Bekkum P, van der Wolf JM, de Vries PM, van Overbeek LS (2000) Survival of Ralstonia solanacearum biovar 2, the causative agent of potato brown rot, in field and microcosm soils in temperate climates. Phytopathology 90:1358–1366
Van Elsas JD, Kastelein P, de Vries PM, van Overbeek LS (2001) Effects of ecological factors on the survival and physiology of Ralstonia solanacearum bv 2 in irrigation water. Can J Microbiol 47:842–854
Van Overbeek LS, Bergervoet JHH, Jacobs FHH, van Elsas JD (2004) The low-temperature-induced viable-but-nonculturable state affects the virulence of Ralstonia solanacearum biovar 2. Phytopathology 94:463–469
Wang YD, Zhao S, Hill CW (1998) Rhs elements comprise three subfamilies which diverged prior to acquisition by Escherichia coli. J Bacteriol 180:4102–4110
Winstead NN, Kelman A (1952) Inoculation techniques for evaluating resistance to Pseudomonas solanacearum. Phytopathology 42:628–634
Zhang YL, Ong CT, Leung KY (2000) Molecular analysis of genetic differences between virulent and avirulent strains of Aeromonas hydrophila isolated from diseased fish. Microbiology 146:999–1009
Acknowledgements
We thank Tracey Timms-Wilson for providing the reference R. solanacearum strain 715, Cartilin Allen for providing UW551 DNA, Christian Boucher for providing access to the genome sequence of strain 1609 prior to online publication, Leo van Overbeek for critically reading this manuscript and helpful discussions and the students Erwin Berendsen and Douwe van der Tuin for technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stevens, P., van Elsas, J.D. A putative genomic island, PGI-1, in Ralstonia solanacearum biovar 2 revealed by subtractive hybridization. Antonie van Leeuwenhoek 98, 359–377 (2010). https://doi.org/10.1007/s10482-010-9450-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-010-9450-4