
Abstract
Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is presenting as a systemic disease associated with vascular inflammation and endothelial injury. Severe forms of SARS-CoV-2 infection induce acute respiratory distress syndrome (ARDS) and there is still an ongoing debate on whether COVID-19 ARDS and its perfusion defect differs from ARDS induced by other causes. Beside pro-inflammatory cytokines (such as interleukin-1 β [IL-1β] or IL-6), several main pathological phenomena have been seen because of endothelial cell (EC) dysfunction: hypercoagulation reflected by fibrin degradation products called D-dimers, micro- and macrothrombosis and pathological angiogenesis. Direct endothelial infection by SARS-CoV-2 is not likely to occur and ACE-2 expression by EC is a matter of debate. Indeed, endothelial damage reported in severely ill patients with COVID-19 could be more likely secondary to infection of neighboring cells and/or a consequence of inflammation. Endotheliopathy could give rise to hypercoagulation by alteration in the levels of different factors such as von Willebrand factor. Other than thrombotic events, pathological angiogenesis is among the recent findings. Overexpression of different proangiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (FGF-2) or placental growth factors (PlGF) have been found in plasma or lung biopsies of COVID-19 patients. Finally, SARS-CoV-2 infection induces an emergency myelopoiesis associated to deregulated immunity and mobilization of endothelial progenitor cells, leading to features of acquired hematological malignancies or cardiovascular disease, which are discussed in this review. Altogether, this review will try to elucidate the pathophysiology of thrombotic complications, pathological angiogenesis and EC dysfunction, allowing better insight in new targets and antithrombotic protocols to better address vascular system dysfunction. Since treating SARS-CoV-2 infection and its potential long-term effects involves targeting the vascular compartment and/or mobilization of immature immune cells, we propose to define COVID-19 and its complications as a systemic vascular acquired hemopathy.
Similar content being viewed by others

Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel RNA virus associated with the outbreak of a coronavirus-associated acute respiratory disease called coronavirus disease-19 (COVID-19) in humans [1]. COVID-19 has been linked to a large range of illness severities, varying from very mild to life threatening. In a cohort of 44,000 people with COVID-19 in China [2], the illness demonstrated three distinct clinical presentations: 81% showed mild symptoms, 14% had severe symptoms including dyspnea, hypoxia and lung parenchymal involvement on computed tomography (CT) scan, and 5% developed respiratory failure and/or multi-organ dysfunction. The overall worldwide case fatality ratio has ranged from 1.8 to 2.3% (www.covid19.who.int). The varied clinical presentations, despite a nearly identical viral genome during the early phases of the pandemic [3], suggests a significant host influence on the clinical manifestations. Interestingly, comorbidities leading to severe COVID-19 disease include age, hypertension, diabetes, ischemic heart disease, vascular disease, renal failure and obesity. Although there is considerable speculation about how these different risk factors contribute to the clinical manifestations of the disease, the pathogenesis of severe COVID-19 and the variable clinical response to the virus remains unclear. Patients with severe COVID-19 not only develop pulmonary disease, eventually culminating in acute respiratory distress syndrome (ARDS) [4, 5], but also display a myriad of extrapulmonary symptoms, including acute kidney injury, acute cardiac injury, coagulopathy, thromboembolic complications, including stroke and pulmonary embolism, and circulatory shock [6, 7]. Together this information suggests that the pathology of COVID-19 has a strong vascular component. In this review, we will summarize current knowledge about hematological and vascular defects associated with COVID-19. In particular, we will describe pathophysiological insights into COVID-19-induced coagulopathy, endotheliopathy and angiogenesis associated defects.
COVID-19-associated coagulopathy
COVID-19 has early been identified as a hypercoagulable and thrombogenic disease, characterized by a high incidence of venous and arterial thrombotic events, in particular in the most severely affected patients [8]. While SARS-CoV-2 is typically characterized by an infection of the upper aerodigestive tract and mild respiratory manifestations, it may progress to severe forms including ARDS and multisystemic disorders. Up to 30% of these critically ill COVID-19 patients admitted to the intensive care unit (ICU) develop thrombotic complications, mainly including pulmonary embolism and deep vein thrombosis, despite pharmacological thromboprophylaxis [9,10,11,12,13]. The detection of SARS-CoV-2 in multiple organs, thrombosis and ischemic complications, and its multisystemic clinical features, have suggested that COVID-19 might be a systemic vascular disease. SARS-CoV-2 respiratory tract invasion is indeed responsible for an intense vascular inflammation with extensive endothelial damage, leading to deregulated coagulation activation and potential subsequent thrombotic manifestations [14,15,16]. Coagulation activation in COVID-19 is a specific feature distinguishing it from other respiratory diseases.
COVID-19 increases the risk of thrombotic events
Pulmonary embolism has been identified as one of the most severe consequences and one of the hallmarks of COVID-19 [17]. In a multicentric prospective cohort, study including 150 patients admitted to the ICU for COVID-19 ARDS, 64 clinically relevant thrombotic complications have been diagnosed, despite at least prophylactic dosing of heparin since the admission in the ICU [9]. Among them, 25 pulmonary embolisms were diagnosed on 100 CT pulmonary angiogram performed because of a respiratory aggravation or a sudden significant increase of the fibrin degradation product D-dimer. Compared to a historic cohort of non-COVID-19 ARDS, COVID-19 ARDS patients developed significantly more thrombotic complications, with an odd ratio of 6.2 [1.6–23.4] (p < 0.01) for pulmonary embolism. COVID-19 patients also displayed many other thrombotic events, like clotting during continuous renal replacement therapy, or of extracorporeal membrane oxygenation device [9].
In a concomitant report, Poissy et al. [13] also noticed an unexpectedly high number of pulmonary embolisms in COVID-19 patients that stay in the ICU (n = 22/107 patients, 20.6%), which was higher than in a historical cohort of ICU patients with influenza virus with similar severity scores on ICU admission (20.6% versus 6.1%; absolute increased risk, 14.4% [95% CI 6.1–22.8]). An increased incidence of arterial thrombosis (mainly stroke and acute coronary syndrome) has also been reported [18, 19]. Several other publications confirmed the high cumulative incidence of thrombotic complications in critically ill patients with COVID-19 admitted to the ICU [10,11,12] and autopsy-based findings stated that thrombotic complications were a major unsuspected cause of death [20]. In a retrospective nested case–control study conducted in two French hospitals, Planquette et al. estimated pulmonary embolism prevalence in COVID-19 patients to be close to 5% in the whole population and nearly 20% of the clinically suspected population. Pulmonary embolism in COVID-19 patients seems to be associated with more extensive lung damage and to require more frequently invasive ventilation [21]. Wichmann et al. have thus diagnosed deep vein thrombosis in more than half of their 12 patients and PE as the direct cause of death in 25% of their patients [22].
Variability in thrombosis prevalence might be explained by nature of different reports. Indeed, most of them are retrospective cohorts with inherent bias, including the absence of systematic screening for thrombotic events, different thromboprophylaxis protocols and a high heterogeneity in patients’ clinical severity. Despite these limitations, several meta-analyses confirmed the high incidence of venous thromboembolism events, mainly for critically ill patients, which were associated with a higher risk of mortality [23,24,25,26], probably through systemic impaired microcirculatory function of organs in most severe forms of COVID-19 with subsequent organ dysfunctions [27]. Most of these reports also accounted for rare severe hemorrhagic complications, suggesting that COVID-19-associated coagulopathy possesses specific characteristics.
Why SARS-CoV-2-associated coagulopathy is unique
The pathophysiology of COVID-19-associated coagulopathy is complex and involves several pathways. It differs from other thrombotic coagulopathies reported in critically ill patients, including disseminated intravascular coagulation (DIC), which is for example commonly encountered in bacterial sepsis.
The respiratory tract invasion by SARS-CoV-2 is responsible for a systemic inflammatory response causing the release of high levels of pro-inflammatory cytokines associated with an adaptive hemostasis response meant to limit the spread of the pathogen, thus constituting the first line of host defense. Pro-inflammatory interleukins (IL)-1β and IL-6, locally produced by the macrophages and monocytes in the lung, may thus induce thrombocytosis and hyperfibrinogenemia. The inflammatory response is therefore responsible for an activation of blood coagulation in almost all COVID-19 patients. Although activation of coagulation is a hallmark of several infectious diseases, the coagulation activation pattern in COVID-19 patients is not the same as in septic patients [9, 28]. Indeed, while sepsis-induced DIC is mainly characterized by low platelet count, prolonged prothrombin time and decreased antithrombin [29,30,31,32], COVID-19 patients display higher fibrinogen levels and increased D-dimers, but minor changes in platelet count, prothrombin time and antithrombin [9]. Furthermore, while 30–40% of septic shock patients develop DIC [28], few COVID-19 patients were diagnosed with DIC according to the International Society on Thrombosis and Hemostasis or the Japanese Association for Acute Medicine diagnosis scores for DIC [9, 13], unless COVID-19 was secondarily complicated by bacterial sepsis, septic shock or multiple organ failure. D-dimer increase has thus been widely assessed in COVID-19 patients and has been correlated to disease severity and mortality [33,34,35,36]. Therefore, monitoring of D-dimers has been proposed by several guidelines but no clear data exist to date on a reliable cut-off to predict mortality risk or to guide the choice of initiating therapeutic anticoagulation [37].
Coagulation activation following this important inflammatory response leads to increased tissue factor (TF) expression, release of neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs) release, hyperfibrinogenemia and increased thrombin generation [38]. The intense vascular inflammation also leads to extensive endothelial damage, characterized by mononuclear cell infiltrate, lymphocytic endothelialitis, platelet activation and subsequent deregulated coagulation activation [14,15,16, 27]. Infection of EC through the receptor angiotensin-converting enzyme 2 (ACE2) is a huge matter of debate since, evidence for ACE2 expression in EC is weak, and the direct endothelial infection route is controversial (as discussed in 3.4.1) The procoagulant phenotype is subsequently enhanced through upregulation of TF expression and decrease of fibrinolysis. Plasmin activity is indeed suppressed by a decrease of urokinase plasminogen activator by alveolar macrophages and an increase of plasminogen activator inhibitor-1 (PAI-1), which will accelerate clot formation in lung microcirculation [39]. Extensive endothelial damage, indirectly assessed by very high levels of von Willebrand factor (VWF) in the circulation, contributes to sustaining the antiviral inflammatory reaction (27, 40). At the onset of the disease, inflammation and coagulation are localized in the lungs [27]. Local endothelial injury may thus favor microvascular clot formation and angiopathy in lungs and other organs, while hypercoagulability along with hyperfibrinogenemia may explain large vessel thrombosis in the systemic circulation when inflammation and coagulation activation become systemic and the disease severity progresses. In severe forms of COVID-19, including hypoxemic pneumonia and ARDS, profound hypoxemia in the pulmonary capillaries may also result in vasoconstriction reducing blood flow, thereby promoting vascular occlusion [41]. Hypoxemia may also induce activation of hypoxia-inducible factors (HIFs), which are heterodimeric transcriptional factors consisting of a HIFβ subunit, expressed by all nucleated cells, and either the HIF1α or the HIF2α subunit (for HIF1 and HIF2, respectively). Hypoxia induces the stabilization of HIF2α subunits by decreasing their hydroxylation, resulting in the induction or inhibition of many genes, including TF and PAI-1, which are released by EC like VWF [42, 43]. Other pathways might also be involved in COVID-19-induced coagulopathy. Previous studies exploring lupus anticoagulant (LA) described between 45 and 88% of positivity in different COVID-19 cohorts in the medical ward and/or ICU settings [10, 23,24,25]. Only one study suggested in vitro that antiphospholipid antibodies (APA) positivity in sera of COVID-19 patients could be prothrombotic but LA testing was not assessed [26]. However, their pathogenicity and relevance need further examination in future studies, as several diseases including infectious ones, may be associated with transient LA, making their clinical relevance in COVID-19 controversial. Gendron et al. investigated the prevalence and prognostic value of conventional and non-conventional APA in COVID-19 patients in a prospective observational French cohort of patients hospitalized for COVID-19 suspicion [44]. 249 patients were hospitalized for suspected COVID-19, including 154 with confirmed COVID-19 and 95 not confirmed. A significant increase in LA positivity among COVID-19 positive patients (60.9% versus 23.7% in non-COVID-19 patients) was observed, while prevalence of conventional (anti-cardiolipin and anti-beta-2-GP1, IgG and IgM isotypes) and non-conventional APA (IgA, anti-phosphatidylserine/prothrombin and anti-prothrombin IgG and IgM) were low in both groups. Positivity for LA in COVID-19 patients was significantly associated with inflammatory biomarkers such as higher fibrinogen and C-reactive protein (CRP) levels. Univariate analysis did not show any association between LA positivity and higher risk of venous thromboembolism events or in-hospital mortality. Unadjusted and adjusted (to CRP, age and sex) Kaplan–Meier survival curves according to LA positivity confirmed the absence of association with venous thromboembolism events or in-hospital mortality [44].
Management of SARS-CoV-2-induced coagulopathy
Increased D-dimer and fibrinogen levels are therefore the most common finding in COVID-19. As other standard coagulation assays are mostly within normal ranges, even in critically ill patients, they are not informative to detect COVID-19-associated procoagulant state [45]. Although D-dimer cut-offs have been associated with poor prognosis in COVID-19 patients, a more comprehensive overview of hemostasis activation taking inflammatory markers into account is necessary. Considering the close interactions between inflammation and coagulation activation, following the kinetics of D-dimers, fibrinogen levels and other inflammatory markers, might help to better manage thromboprophylaxis in these patients. Ranucci et al. [46] thus reported the association between the pro-inflammatory cytokine IL-6 and fibrinogen levels in COVID-19 ARDS. Assays to assess global coagulation status include thrombin generation testing, thromboelastography and rotational thromboelastometry. These assays respectively showed increased thrombin generation and procoagulant patterns with increased clot strength in COVID-19 patients despite at least prophylactic anticoagulation, consistent with high fibrinogen levels [46,47,48,49,50]. In addition to hyperfibrinogenemia-related hypercoagulability, impaired fibrinolysis could contribute to the severe coagulopathy observed in these patients [51]. Fibrin formation will itself trigger activation of fibrinolysis, by binding tissue-type plasminogen activator and plasminogen to generate plasmin. Urokinase-type plasminogen activator and its receptor, which are predominantly extravascular and are known to be active on lung epithelia, may also contribute [52]. Plasminogen activator inhibitor 1 (PAI-1), the main inhibitor of tissue plasminogen activator and urokinase-type plasminogen activator, is expressed by ECs, epithelial cells, monocytes and macrophages [53]. Endothelial and platelet activation results in the release of PAI-1 and downregulates fibrinolysis [54]. Elevated expression of PAI-1 facilitates tissue fibrosis by inhibiting plasmin-mediated activation of tissue matrix metalloproteases and plasmin degradation of misfolded protein [55] and in this way may contribute to the emerging problem of long-term complications in COVID-19 survivors [56]. Wright et al. reported a lack of fibrinolytic activity in 57% of their patients and showed it was a predictor of venous thromboembolism events [57]. This phenomenon, described as ‘fibrinolysis shutdown’, has been shown to be a predictor of first and recurrent pulmonary embolism, micro- and macrovascular thrombosis [58] and potentially alveolar fibrin deposition [59, 60]. Kruse et al. also found reduced fibrinolysis in COVID-19 patients at the ICU and showed it was associated with thromboembolism [61].
According to the second version of the International Society on Thrombosis and Hemostasis recommendations [62], and the French recommendations [37], prophylactic dosing of heparin might be intensified if patients require critical care support and if there are no contraindications. Furthermore, therapeutic anticoagulation should be administered if there is any evidence of thrombotic event or if pulmonary embolism is highly suspected and an imaging examination is not feasible to confirm the diagnosis. Because the combination of numerous recent reports on thrombotic complications in critically ill COVID-19 patients and observational ones suggesting that aiming for higher anticoagulation targets for thromboprophylaxis of severe COVID-19 patients could decrease the rate of thrombosis [63], several paradigms have been proposed for anticoagulation of COVID-19 patients according to clinical severity. Higher anticoagulation targets were considered to protect from thrombotic events in the most severely affected patients [62, 37]. Helms et al. demonstrated that anticoagulation was not associated with increased bleeding, differences in ICU mortality or length of stay compared with patients in regular prophylaxis [64, 65]. In an observational retrospective study, Paranjpe et al. [66] assessed the association between administration of in-hospital anticoagulation and survival in a large cohort of hospitalized patients with COVID-19 and showed that therapeutic anticoagulation may be associated with improved outcomes among patients hospitalized with COVID-19 (reduced risk of mortality: adjusted HR of 0.86 per day, 95% confidence interval 0.82–0.89, p < 0.001). In another observational retrospective study, Chocron et al. aimed to determine whether anticoagulation therapy modifies the risk of developing severe COVID-19. Patients with COVID-19 initially admitted in medical wards of 24 French hospitals were included prospectively from February 26 to April 20, 2020. The study enrolled 2878 patients with COVID-19, among whom 382 (13.2%) were treated with oral anticoagulation therapy before hospitalization. After adjustment, anticoagulation therapy before hospitalization was associated with a better prognosis with an adjusted hazard ratio of 0.70 (95% CI 0.55–0.88). In contrast, therapeutic or prophylactic low or high dose anticoagulation started during hospitalization were not associated with any of the outcomes. These results are in line with an improvement of disease when anticoagulation therapy is introduced in early disease. Several randomized studies seem to confirm this hypothesis (https://www.remapcap.org/protocol-documents), that efficient high dose anticoagulation therapy only determines COVID-19 prognosis in moderate disease but not in the severe form, in which prophylactic treatment is the best way to avoid hemorrhage, but it does not add any benefit in terms of disease outcome. Regarding the use of an anticoagulation regimen, a recently published randomized study, including COVID-19 patients admitted to the ICU, intermediate-dose prophylactic anticoagulation compared with standard-dose prophylactic anticoagulation did not result in a significant difference in the primary outcome of a composite of adjudicated venous or arterial thrombosis, treatment with extracorporeal membrane oxygenation, or mortality within 30 days [67]. These results are not in favor of a routine use of intermediate-dose prophylactic anticoagulation in unselected ICU-admitted COVID-19 patients. Thus, the benefit-risk balance of higher anticoagulation targets should take the risk of bleeding events into account and previous recommendations should be amended in line with these new data.
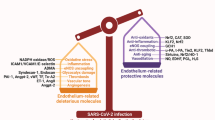
COVID-19 is an endothelial disease
EC have been considered as a passive inner lining for so many years. However, the vascular endothelium is now perceived as an independent and important organ system that is centrally involved in hemostatic balance, cellular trafficking, regulation of vascular tone and the passage of fluids [68]. It has been recognized that global endothelial dysfunction, in particular breakdown of the vascular barrier, represents a cornerstone in the development of multi-organ failure in sepsis [69]. The endothelium is, in turn, shielded against pathogenic insults by a negatively charged, gel-like mesh consisting of highly sulfated glycosaminoglycans and proteoglycans—the so-called endothelial glycocalyx (eGC) [70, 71]. Thinning of this up to 3 μm thick eGC plays a causative role in leukocyte recruitment, hyperpermeability and the development of end-organ damage, in particular ARDS and acute kidney injury (AKI) [71, 72]. In this chapter, we attempt to present an overview of the markers and potential mechanism of endothelial dysfunction in COVID-19. In addition, we provide support for the hypothesis that COVID-19 is indeed a vascular disease.
Visualization of endothelial dysfunction in sublingual microvessels
Among the numerous methods for quantifying endothelial dysfunction, in vivo, intravital imaging of the sublingual microcirculation probably provides the most vivid evidence. Pilot studies using hand-held videomicroscopy revealed only a small reduction in total and perfused vascular density as well as evidence of microthrombosis in sublingual microvessels of mechanically ventilated COVID-19 patients (some on extracorporeal membrane oxygenation) [73, 74]. Rovas et al. used a novel state-of-the-art image acquisition and analysis approach to quantify even subtle alterations of the sublingual microcirculation. COVID-19 patients (n = 23) showed an up to 90% reduction in vascular density, that was almost exclusively limited to small capillaries (diameter 4–6 µm), and a significant reduction in capillary red blood cell velocity. Capillary impairment correlated with the sequential organ failure assessment (SOFA) and sepsis-induced coagulopathy (SIC) score as well as the oxygenation index, indicating that sublingual capillaries are, at least in part, representative of the pulmonary and systemic microvasculature [75]. All videomicroscopy studies reported a strong association between D-dimer level and microvascular alterations, thus indicating that capillary clogging by fibrinous microthrombi, which has already been shown by autopsy studies in lungs from COVID-19 patients [27, 76], plays certainly an important role here [73,74,75].
Damage of the endothelial glycocalyx in COVID-19
Damage of the eGC is considered an early feature of endothelial dysfunction in various diseases [70]. The dynamic lateral movement of red blood cells into the permeable part of the eGC layer, expressed as the perfused boundary region (PBR, in µm), provides a robust estimate of the glycocalyx thickness in sublingual microvessels [77,78,79]. Especially COVID-19 patients on mechanical ventilation showed severe glycocalyx damage as indicated by higher PBR values (i.e., thinner glycocalyx) and increased blood levels of shed glycocalyx constituents (such as Syndecan-1). Of note, the PBR value showed the best discriminatory ability to predict 60-day mortality in a cohort of COVID-19 patients [75]. Damage to the eGC is probably not specific to certain eGC constituents, as numerous glycosaminoglycans and their fragments were significantly elevated in the blood of COVID-19 patients [72, 80, 81].
Stahl et al. also found significantly increased PBR values and increased syndecan-1 levels in 19 mechanically ventilated COVID-19 patients. Furthermore, they were able to show that, unlike in bacterial sepsis, it was not the eGC damaging enzyme heparanase-1 that was upregulated, but that its intrinsic inhibitor, heparanase-2, was significantly reduced. Concomitant in vitro experiments showed that EC stimulated with COVID-19 serum showed lower transcription of heparanase-2 and less eGC coverage. Transgenic overexpression of heparanase-2 in a lentivirus-transduced EC line was sufficient to reverse this phenotype. The authors therefore postulate that acquired heparanase-2 deficiency might represent a potential mechanism of injury to the eGC, which could eventually progress to widespread endothelial dysfunction in COVID-19 [80]. However, using a more sophisticated assay, Buijsers et al. found elevated plasma heparanase-1 activity in COVID-19 patients (n = 48), that correlated with cleaved heparan sulphate levels and clinical markers of disease activity. Treatment of EC with low molecular weight heparin (LMWH), which serves as an alternative substrate for heparanase and thus acts as a dose-dependent heparanase inhibitor, prevented glycocalyx perturbation induced by plasma from COVID-19 patients [82]. Interestingly, heparanase activity was significantly lower in patients with prophylactic low-molecular weight heparin (LMWH) treatment [83]. Therefore it would be quite conceivable, that the 40% lower risk of death in patients receiving LMWH or unfractionated heparin, which was recently reported by an Italian multicenter study, is partly due to the protection of the eGC [84]. Given that the SARS-CoV-2 spike glycoprotein (S protein) contains glycocsaminoglycan (GAG)-binding-like motifs, further studies are needed to clarify if GAG remodeling, as seen in e.g. diabetes [85], amplifies viral host cell entry [86].
The Angiopoietin/Tie2 ligand–receptor system
Local formation of microthrombi and edema in the lungs, the hallmarks of ARDS in COVID-19 patients, requires a switch of the endothelial phenotype from a quiescent towards a pro-adhesive, pro-inflammatory, hyperpermeable activated state. This process is non-redundantly controlled by Tie2, a receptor that is highly enriched in the endothelium and actively signals vascular quiescence [87]. Under physiological conditions, Tie2 is tonically activated by angiopoietin-1, a vasculoprotective protein secreted by pericytes and platelets [88]. In bacterial sepsis, its intrinsic antagonist called angiopoietin-2 is rapidly released from activated endothelium, competitively inhibits Tie2 while activating certain integrins, leading to endothelial destabilization [89, 90], and is a biomarker that predicts mortality [91,92,93]. In a cross-sectional study, Rovas et al. found that angiopoietin-2 was already increased in non-ventilated SARS-CoV-2 infected patients, indicating that angiopoietin-2 may unleash endothelial inflammation in COVID-19 early on [75]. In this regard, Smadja et al. measured angiopoietin-2 in 40 consecutive COVID-19 patients admitted to the emergency department with need for hospitalization [94]. Angiopoietin-2 admission levels were significantly associated with D-dimer, CRP and creatinine levels. Furthermore, an angiopoietin-2 cut-off of 5.0 ng/mL (normal range usually < 1 ng/mL) was identified as best predictor for ICU admission. In patients on mechanical ventilation, angiopoietin-2 correlated inversely with pulmonary compliance, a measure of the lung's ability to stretch and expand [94]. Another group determined angiopoietin-2 levels on ICU admission in 38 COVID-19 patients, most of whom were mechanically ventilated [95]. Angiopoietin-2 was significantly higher in non-survivors (n = 10) and a quite similar cut-off of 4.0 ng/mL predicted ICU mortality with a sensitivity of 89% and a specificity of 77%. Interestingly, the levels of the protective angiopoietin-1 were the same in both groups, a finding that is consistent with two other reports [75, 94] and many previous studies on ARDS in bacterial sepsis [96]. Bermejo-Martin et al. analyzed the viral RNA load of SARS-CoV-2 (RNAemia) in plasma samples of 250 COVID-19 patients (50 outpatients, 100 hospitalized ward patients and 100 critically ill). Consistent with previous report, angiopoietin-2 levels were highest in critically ill patients and beyond that correlated tightly with viral RNAemia, which had a frequency of almost 80% in the ICU group [97]. Pine et al. compared the performance of 16 biomarkers of angiogenesis and endotheliopathy in 49 hospitalized COVID-19 patients. The level of angiopoietin-2 increased with disease severity and was the second-best predictor of in-hospital mortality after follistatin [98]. Taken together, available data are very similar to what we know about angiopoietin-2 from sepsis studies. Fortunately, numerous preclinical models have clearly shown that Tie2-activating therapeutics can effectively prevent sepsis-induced ARDS [96]. The angiopoietin/Tie2 system is therefore a very promising target for future studies on the prevention and treatment of ARDS in COVID-19.
The Vascular Endothelial Growth Factor signaling axis
The vascular endothelial growth factor (VEGF) family and its receptors are essential regulators of angiogenesis and barrier function. Currently, the VEGF family consists of VEGF-A, PlGF, VEGF-B, VEGF-C, VEGF-D, VEGF-E. Several investigators found elevated plasma levels of VEGF-A in sera of COVID-19 patients, which correlated with disease severity [75, 97,98,99]. While VEGF-B levels appeared to be unchanged in COVID-19, VEGF-D, which promotes angiogenesis and lymphangiogenesis [100] was lower in COVID-19 compared to healthy controls and correlated inversely with the sequential organ failure assessment (SOFA) score [75, 98]. In contrast, Kong et al. identified elevated VEGF-D as the most important indicator of disease severity, outperforming D-dimer, IL-6 levels and lymphocyte count, in a small cohort of COVID-19 patients [101]. Further studies are needed to clarify these contrasting findings and the potential mechanistic role of VEGF-D in COVID-19. Recently, VEGF-A, PlGF and FGF-2 were quantified and examined for their association with in-hospital mortality of adult COVID-19 patients [99] in consecutive ambulatory and hospitalized patients in the first 48 h following admission. It was found that levels of VEGF-A, PlGF and FGF-2 significantly increase with severity of the disease (p < 0.001). PlGF levels above 30 pg/mL was identified as the best predictor of in-hospital mortality in COVID-19 patients. Survival analysis after stratification based on PlGF expression confirmed its value for in-hospital mortality prediction. This result was found using Kaplan–Meier survival curves (p = 0.001) and a Cox proportional hazard model adjusted to age, body mass index, D-dimer and CRP levels (3.23 95% CI [1.29–8.11], p = 0.001). The levels of angiogenesis markers in COVID-19 was associated with the presence of intussusceptive angiogenesis observed in lung tissue of COVID-19 patients [102]. This could be an argument for testing antiangiogenic strategies [103] as a new interesting therapeutic approach in COVID-19.
Soluble levels of Flt-1 (sFlt-1), a circulating truncated form of the VEGF-A receptor Flt-1/VEGFR1, were markedly increased in COVID-19 patients and correlated with disease severity [75, 98]. Under normal conditions, sFlt-1 binds electrostatically to proteoglycans and is thus sequestered within the eGC [104]. Elevated sFlt-1 could therefore theoretically also be a consequence of the eGC damage. Overexpression of sFlt-1 has been well demonstrated to promote endothelial dysfunction, most notably during preeclampsia [105]. Dupont et al. reported that sFlt-1 plasma levels at ICU admission (n = 46) were associated with the need for mechanical ventilation, the need for vasopressor support, development of severe acute kidney injury and death [106]. In contrast to preeclampsia, however, elevated sFlt-1 levels in COVID-19 are clearly not accompanied by a reduction in PlGF [107, 108]. This finding is apparently very consistent, as the sFlt-1/PlGF ratio remains low in pregnant women with COVID-19 pneumonia, thus allowing a good differentiation between true preeclampsia and preeclampsia-like symptoms due to COVID-19 [109].
Vascular adhesion molecules
Vascular adhesion molecules expressed on activated endothelium are known to bind to leukocyte integrins during inflammation and promote immune responses. They also exist in soluble forms in human plasma, due to activation and proteolysis mechanisms at cell surfaces. Bermejo-Martin et al. analyzed soluble intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 in 250 patients with COVID-19 [50 outpatients, 100 hospitalized ward patients and 100 critically ill]. Especially VCAM-1 but also ICAM-1 levels correlated with SARS-CoV-2 RNAemia [97]. Orfanos et al. found that soluble E-selectin and soluble ICAM-1 correlated with disease severity and predicted ICU mortality in 38 critically ill COVID-19 patients [95]. Similarly, serum levels of soluble platelet endothelial adhesion molecule (sPECAM-1) were elevated and correlated with disease severity in COVID-19 patients (n = 38), whereas asymptomatic carriers had similar sPECAM-1 levels as healthy controls [110]. In a cohort of 100 randomly selected hospitalized COVID-19 patients, plasma P-selectin levels were independently associated with a composite outcome, based on occurrence of thrombotic events and death [111]. Smadja et al. analyzed E-selectin in 40 consecutive COVID-19 patients admitted to the emergency department with need for hospitalization. Both, gene expression and soluble E-selectin protein increased with severity in a grade-dependent manner and were significantly higher in patients admitted to the ICU [94]. Endothelial dysfunction, and in particular E-selectin levels, have also been associated with SARS-CoV-2-related multisystem inflammatory syndrome in children (MIS-C) with shock, in particular with the vasoactive and inotropic score. These results highlight the significant relationships between endothelial dysfunction, systemic hyper-inflammation, and acute severe cardiovascular manifestations. Endothelial dysfunction may be one of the mechanisms underlying SARS-CoV-2-related MIS-C with shock [112].
The von Willebrand Factor-ADAMTS13 axis
VWF is a multimeric glycoprotein mainly synthesized by EC and stored in Weibel-Palade bodies, the storage granules of ECs. Importantly, Weibel-Palade bodies also contain angiopoietin-2, and the exocytosis of these compartments, leading to release of angiopoietin-2 and VWF, is triggered by EC agonists such as VEGF and histamine [113]. Therefore, Weibel-Palade body release from ECs constitutes a link between inflammation and coagulation, and the observation that both VWF and angiopoietin-2 levels are increased in COVID-19 patients suggests that Weibel-Palade body exocytosis upon EC activation could be instrumental in the disease progression of COVID-19. As VWF is heavily involved in platelet aggregation and thrombus formation following endothelial activation, it has been studied extensively as a potential biomarker of COVID-19 severity and a potential risk factor for COVID-19 related death. Several retrospective studies have reported elevated VWF levels in COVID-19 patients, the highest levels being found in critically ill patients. Early in the COVID-19 pandemic, Panigada et al. reported strongly elevated VWF levels in a cohort of 24 COVID-19 patients treated in ICU [49]. In addition, Goshua et al. found in 68 patients of varying clinical severity COVID-19 patients that although nearly all patients displayed increased VWF levels, significantly higher levels were found in critical ICU patients [14]. Furthermore, Philippe et al. demonstrated that VWF levels measured at the time of admission in a cohort of 208 COVID-19 patients, ranging from outpatients to critically ill, showed to be a powerful predictor of in-hospital mortality, advocating for a role of VWF in promoting harmful microthrombosis in COVID-19. Specifically, a threshold of 423% of VWF (normal values: 50–150%) offered an optimal sensitivity–specificity balance [114]. Both Vassiliou et al. and von Meijenfeldt et al. confirmed this observation by reporting from cohorts of, respectively, 38 ICU and 102 mixed-severity COVID-19 patients, a significantly higher level of VWF factor at admission in non-survivors than in survivors [95, 115].
Taken together, these data suggest that VWF elevation is an accurate mirror of the intensity of endothelial damage, most likely resulting from a dysregulated immune-inflammatory response, as shown by the strong association between VWF levels and circulatory inflammatory cytokines in COVID-19 patients in the ICU [116]. On its own, such a significant increase of VWF could explain its prothrombotic effect but additional qualitative anomalies of VWF have been described in COVID-19.
Physiologically, ECs release VWF in the bloodstream in the form of hyper-reactive ultra-large VWF multimers, which remain tethered to the EC surface. Upon release, ultra-large VWF strands are cleaved by the plasma protease A Disintegrin And Metalloproteinase with ThromboSpondin motifs (ADAMTS)13 into smaller multimers, which ensure VWF hemostatic function without harmful/negative thrombotic effects. Several authors described a moderate decrease in circulating ADAMTS13 levels in COVID-19 patients, being more pronounced in critically ill patients [117,118,119,120]. Interestingly, an association between ADAMTS13 decrease and vascular density in sublingual microvessels of mechanically ventilated COVID-19 patients was found, in favor of a link between microthrombosis and reduced ADAMTS13 function [75]. Philippe et al. suggested an ADAMTS13 ‘overflow’ due to a major quantitative imbalance between the substrate and the enzyme, and reported an eightfold increased VWF-to-ADAMTS13 activity ratio in COVID-19 patients that required high-intensity care and mechanical ventilation [114]. Despite being time-consuming, assessment of the VWF multimeric profile using gel electrophoresis is valuable as a direct readout of VWF functionality, with a greater proportion of high molecular weight multimers (HMWM). Indeed the levels of VWF HMWM were significantly increased in critically ill compared to non-critically ill COVID-19 patients [114]. In addition, patients with an excess of HMWM displayed abnormally high VWF-collagen binding capacity. Accordingly, Turecek et al. reported abnormal persistence of highly thrombogenic ultra-high-molecular-weight VWF multimers in a cohort of 36 severely ill COVID-19 patients. Moreover, in vitro incubation of plasma samples from patients with recombinant ADAMTS13 substantially reduced VWF HMWM in a time- and concentration-dependent manner [121]. In contrast, Doevelaar et al. reported decreased VWF HMWM in 75 patients with confirmed COVID-19 of mild to critical severity [122], an observation shared by Mancini et al. in a cohort of 50 hospitalized COVID-19 patients [123]. The reason for such discrepancy between studies is not obvious. Both the use of extracorporeal membrane oxygenation in intensive care and the consumption of larger VWF multimers in microthrombi could constitute potential explanations (Fig. 1). Future studies will have to clarify this point, for example by longitudinal monitoring of VWF multimeric profile throughout hospitalization in a large cohort of COVID-19 patients.

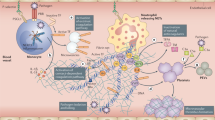
Pathophysiology for microthrombosis in patients with COVID-19. The figure summarizes hypothetical steps of the thrombotic sequence from direct or indirect of SARS-CoV-2 effects on endothelial cells inducing an endotheliopathy and a coagulopathy leading to lung obstruction and potential consequences on the right heart ventricle
Thus, despite this already large number of studies, some issues remain to be clarified regarding VWF involvement in COVID-19 pathophysiology.
Microthrombosis and mechanisms of vascular damage in COVID-19
Vascular and endothelial heterogeneity
The microvasculature comprises the body’s network of arterioles, capillaries and venules. In terms of control of the hemostatic balance, they differ from larger vessels in several important characteristics [124]. Firstly, in the presence of inflammation, locally released cytokines and procoagulant surface molecules such as TF reach very high concentrations in the microvasculature. Secondly, smaller vessels are exposed to higher shear rate and stress. Thirdly, microvascular beds can have specific features, such as the low permeability of the blood–brain barrier and the filtering function of the glomerulus. Finally, capillaries are the site of oxygen exchange.
ECs from different microvascular beds have unique phenotypes, functions, and molecular signatures. These differences reflect tissue-specific molecular signatures, which are now being characterized at the genomic level [125]. The heterogeneity also applies to regulators of hemostasis and thrombosis. For example, VWF is expressed in many but not all microvascular beds [126,127,128], being particularly high in lung and brain ECs but absent from liver sinusoidal endothelium. In animal models, inflammatory stimuli have been shown to regulate VWF expression differently in different organs [129]. These mechanisms may help to explain the high frequency of microthrombi found preferentially in the lungs of patients with systemic inflammation, including patients with COVID-19.
Clinical evidence of microthrombosis in COVID-19
Microvascular thrombosis is a key feature of several disorders with high mortality, such as cerebral malaria and sepsis and is closely associated with organ dysfunction and multi-organ failure. The term microthrombosis indicates the presence of thrombosis in the microvasculature and generally implies that the thrombosis has arisen in situ rather than having arrived as an embolus from a distal venous thrombus. This distinction is particularly important in COVID-19, where a high frequency of thrombi identified in the lungs have been reported as pulmonary embolism but without the expected corresponding prevalence of venous thrombotic events [130]. Manifest infection and inflammation in the lungs with associated hypoxia provide a plausible mechanism for the local generation of microvascular thrombi via EC activation. However, these observations provide only circumstantial evidence.
Post-mortem examination of the lungs of COVID-19 patients could help confirm the hypothesis of microvascular thrombosis in situ, but this has unfortunately been limited due to infection risk and thus direct evidence is scarce. In a series of 18 post-mortem analyses, pulmonary microthrombi were the most noted feature, present in 77.8% of cases, but hemorrhage and alveolar fibrin deposits were also common (50% each). Thus, the increased levels of D-dimers observed in patient plasma may originate from thrombosis, hemorrhage or alveolar exudates, making interpretation difficult and significance obscure [131]. Similarly, post-mortem analysis in an Italian series of COVID-19 patients described platelet–fibrin microthrombi in small arterial (< 1 mm diameter) vessels of the lungs in 33/38 (87%) of cases [132]. In some studies, microvascular thrombosis was less common and larger thrombi were observed [15]. Microvascular thrombi in the lungs of COVID-19 cases were found to be ninefold more frequent than in influenza virus cases [133]. Based on the distribution of thrombi in small vessels with complete occlusion, Lax et al. concluded that the thrombi arose in situ [134].
The formation of microvascular thrombi, in addition to the typical alveolar pathology of pneumonia, provides a good explanation for the sometimes sudden development of hypoxemia in COVID-19 patients, because the thrombi prevent gas exchange in the oxygenated areas of the lung. A similar effect, however, can also arise from large vessel thrombi and typical pulmonary emboli, which are undoubtedly present, as previously discussed [135, 136]. Early in the pandemic, ICU physicians suspected the presence of vascular occlusions based on changes in lung resistance and oxygen transfer, before they were subsequently identified by imaging [137]. Sophisticated imaging techniques such as dual-energy CT (DECT) scanning, which can delineate the perfusion of the lung, have supported this hypothesis. In the largest series of DECT so far, Patel et al. found evidence of widespread blood vessel abnormalities often distinct from areas of infection and the presence of dilated vessels, previously described in pulmonary tumor thrombotic microangiopathy [138]. Other DECT studies have also shown loss of perfusion restricted to consolidated areas [139].
Pathways involved in microvascular thrombosis in COVID-19
Given the hypothesis that thrombi form in situ, the composition of the thrombus in patients with COVID-19 is of great interest, but has not yet been studied in detail, and most reports refer simply to fibrinous or fibrin-platelet thrombi [132, 140]. Despite this limitation, the pathways discussed below have been implicated, based on direct and indirect evidence including circulating plasma levels of the molecules involved. As described above, VWF released following endothelial activation, is greatly elevated in plasma from COVID-19 patients [114, 14, 114]. These very high plasma levels are consistent with endothelial activation, could drive the generation of thrombi rich in VWF, and captured platelets. Ultra-large multimers of VWF associated with a slight decrease of ADAMTS13 levels or function may induce features reminiscent of thrombotic thrombocytopenic purpura (TTP). Remarkably, the blood group ABO locus, which is a strong quantitative trait locus for VWF, was one of only two loci associated with respiratory failure in COVID-19 [141, 142]. Strikingly, non-group-O patients, who are known to have lower levels of VWF and low risk of venous thromboembolism events, also exhibit a low risk of severe COVID-19 when compared to group-O patients [142]. Moreover, SARS-CoV-2 has been described to directly interact with the blood group A antigen expressed on respiratory epithelial cells providing proof of association between SARS-CoV-2 and the ABO (H) genetic locus [143].
The expected consequence of VWF release from ECs is capture of platelets, which have been observed prominently in COVID-19 thrombi, secondary to this is the capture and activation of leukocytes. Leppkes et al. examined autopsy-derived lung tissues from COVID-19 patients and found that the clots in the microvasculature were cell-rich with evidence of NETS [144]. NETS are highly thrombotic structures formed when neutrophils, driven by inflammation, unravel their DNA and extrude it from the cell. Histones from the extruded DNA are potent platelet activators and DNA provides a negatively charged surface, inducing the contact activation system. The degradation of fibrin-rich thrombi is driven by the protease plasmin, which is also involved in the cleavage of viruses’ envelope proteins, including those of other SARS viruses [145]. In addition to the formation of fibrin thrombi, ARDS is characterized by increased alveolar capillary permeability and exudation into the alveoli of fluid rich in inflammatory cells, pro-inflammatory cytokines such as IL-6 and tumor necrosis factor (TNF)-α and coagulation factors including fibrinogen [146, 147]. This leads to fibrin deposition in the air spaces and lung parenchyma, as seen in patients with COVID-19 [148]. Potential abnormalities in fibrinolysis could also participate to microthrombosis and have been described in the first chapter of this review.
Mechanisms of endothelial activation and damage in COVID-19: direct vs indirect
Two main mechanisms have been proposed to explain endothelial activation and damage in patients with COVID-19: direct infection of ECs by SARS-CoV-2 and indirect damage due to circulating mediators, including cytokines, complement activators, immune cells and/or activated platelets.
Direct SARS-CoV-2 infection of EC
Evidence of direct infection of EC by SARS-Cov2 is controversial. The concept was popularized by histopathological features of severe COVID-19 infections. In April 2020, Varga et al. [149] reported the presence of dense particles by electron microscopy (EM) in the post-mortem kidney tissue from one patient. These particles were interpreted as viral inclusion structures in ECs. No immunostaining was carried out to confirm that the structures were indeed viral particles. In the same study, H&E staining of the lung and small intestine from two other patients showed mononuclear and neutrophilic infiltration. Although no direct evidence of endothelial activation was shown, the findings were interpreted as evidence of “endothelialitis” in these tissues. EM findings as evidence of viral infection in ECs have since been disputed by Goldsmith et al. [150]. However, neither study actually reports direct evidence of the presence of viral particles in ECs. A subsequent study on autopsies from all major organ systems suggested that rare coronavirus-like particles, observed by EM, were only present in the ECs of the kidney. Also, in this study immunohistochemistry could not identify the virus in kidney ECs [15]. These authors did not observe EC infection in other organs, nor histological evidence of endothelialitis [15]. Thus, the evidence of direct infection of the endothelium by SARS-CoV2 in post-mortem tissues is limited and inconclusive.
Experimental evidence is also contradictory. In support of SARS-CoV2 binding to ECs is a study by Monteil et al. [151], using engineered human blood vessel organoids from induced pluripotent stem cells (iPSCs), which produced CD31+ endothelial-like networks with pericyte coverage. Here, the presence of the virus in the organoids was shown as evidence of infection and replication in ECs, but the evidence from this study does not rule out that the infection might instead have occurred in pericytes, which also express ACE2 [152, 153]. In contrast, a recent study investigating direct infection of SARS-CoV2 in lung and cardiac ECs in vitro found very low SARS-CoV-2 replication levels [153]. More studies will be required to confirm whether SARS-CoV2 can indeed infect ECs and whether these cells support viral replication.
Expression of ACE2 in endothelial cells
For direct viral infection of ECs, the receptors for the virus should be expressed on EC. The evidence for expression of ACE2, the main receptor for SARS-CoV2, in ECs is weak. Analysis of 13 datasets on the ENCODE database found very low or no ACE2 expression in human ECs from various origins, compared to epithelial cells [153]. Two separate single-cell RNAseq (scRNAseq) studies of the human heart revealed that pericytes, unlike ECs, express ACE2 [152, 153]. Despite this limited evidence, a multitude of review articles has been published stating that ACE2 is expressed on EC, and the model of direct infection is based on this. Most papers cite an immunohistochemistry study of human tissues showing ACE2-positive vessels [154]. However, the ACE2-positive cells observed in this study could be pericytes rather than ECs. Whether ACE2 expression is upregulated in lung ECs of COVID-19 patients is not clear. Ackerman et al. report a significant increase in ACE2 expression in epithelial cells and ECs in lungs from COVID-19 and influenza post-mortem tissues, comparted to controls, as measured by immunohistochemistry [102]. Again, the possible confounding role of pericytes in this finding is unclear. Ongoing studies from the human cell atlas consortium and others will soon provide the answer to this crucial question. Until then, models of SARS-CoV2 damage of ECs should consider direct infection via ACE2 as a hypothesis to be confirmed. Finally, ACE2 is not the only possible endothelial receptor for viral entry: other molecules including neuropilin-1, as well as integrings, have been suggested as possible receptors for the virus as well [155–158], but their role in supporting SARS-CoV-2 infection of ECs remains to be demonstrated.
Indirect activation of EC by COVID-19
While evidence for direct virus infection of EC is debatable, several lines of evidence support the notion that endothelial damage reported in severely ill COVID-19 patients is secondary to infection of neighboring cells, circulating pro-inflammatory cytokines, immune cells, platelets and complement activation. Numerous studies have profiled the highly inflammatory content of plasma of patients with severe COVID-19 and identified some candidate biomarkers of outcome. Among these are powerful activators of the endothelium, such as IL-6, TNF-α, IL-1, interferon-γ, angiopoietin-2 and many others [6, 159, 94]. Many of these factors are known to destabilize the endothelium and/or cause increased endothelial permeability or hyperpermeability, and are likely to disrupt endothelial barrier function in COVID-19, particularly in conjunction with factors such as VEGF and/or thrombin. Endothelial barrier function is essential to prevent excessive fluid loss leading to edema, which is a hallmark of severe COVID-19, and is regulated by intercellular adhesion complexes formed by vascular endothelial (VE)-cadherin [160]. In addition, endothelial monolayer integrity and barrier function are strongly regulated by integrin-dependent extracellular matrix adhesion [90, 161, 162]. Severe disruption of endothelial cell adhesion, and/or the initiation of other signaling pathways by inflammatory agents may not only induce exaggerated permeability but may also result in EC apoptosis and detachment. Direct evidence that plasma from critically ill and convalescent patients with COVID-19 causes EC cytotoxicity was recently reported by Rauch et al. [163]. Thus, the concept of a systemic inflammatory response secondary to lung injury, which can activate and injure the endothelium, as well as contribute to the microthrombotic phenotype is widely accepted [164, 165].
Therapeutic implications
Etiology of the greatly increased frequency of thrombosis in COVID-19 is likely to have important implications for therapy. Conversely, the efficacy of different therapies can be used to deduce the responsible mechanisms.
Several observational studies have reported benefit from increased intensity of standard anticoagulant therapy with heparin [166, 167]. Most recently, the combined platform trials have reported that use of therapeutic anticoagulation in pre-ICU patients resulted in a reduction in the primary outcome of ‘organ support free days’. This was not the case in more severely affected patients already in the ICU receiving organ support (https://tinyurl.com/dox4kgjf).
This finding supports the conclusion that thrombin generation is an important step in thrombus formation in COVID-19 patients and is consistent with the evidence of fibrin-containing thrombi described in post-mortem tissues. We demonstrated that COVID-19-positive patients treated with therapeutic anticoagulation prior to admission had fewer circulating ECs than those without [35]. Since circulating ECs have been described as one of the best circulating vascular integrity biomarkers [168], anticoagulation could be a good therapeutic beyond treating COVID-19-associated coagulopathy but also a good preventive way to avoid endothelial lesion associated with COVID-19. This hypothesis seems correct at least in some populations hospitalized with COVID-19 [169].
As noted above, the contribution of changes in fibrinolytic activity to microthrombosis remains unclear and plasma measurements may not reflect activity in the pulmonary vasculature or the alveoli [170]. Nonetheless, if the thrombi contain substantial amounts of fibrin, as seen in post-mortem samples, enhancement of endogenous fibrinolytic systems may be beneficial. Indeed, intravenous and inhaled tissue plasminogen activator (tPA) have both been reported as beneficial in COVID-19 [168,47,170,173].
Although the microthrombi in COVID-19 are not usually associated with the mechanical destruction of red cells and the reduction in platelet counts seen in thrombotic thrombocytopenic purpura, their enrichment in VWF opens other potential therapeutic opportunities. Recombinant ADAMTS13 (rADAMTS13) has been developed for the treatment of thrombotic thrombocytopenic purpura and has been shown to be effective in degradation of thrombi in animal models of stroke [174]. Thus, a trial of rADAMTS13 in COVID-19 may be both clinically beneficial and a useful confirmation of the microthrombus hypothesis [175]. Recent studies have also reported benefit from the use of antiplatelet agents [176].
Another attractive approach to hamper excessive VWF large multimers binding to platelets and the ensuing microthrombosis could be the use of novel thrombotic thrombocytopenic purpura treatments caplacizumab [177] or anfibatide [178], both of which inhibit binding of the platelet receptor GPIX-Ib to VWF. Moreover, treatment with N-acetyl cysteine, known to reduce the size of VWF multimers, could also be a useful, easy, and cheap strategy in COVID-19 [179].
In addition to strategies targeting the formation of thrombi, the clear involvement of endothelial hyperpermeability and/or endothelial damage, opens several interesting opportunities for therapeutic intervention in critically ill COVID-19 patients, as disruption of the pulmonary microvasculature leading to tissue edema is a major manifestation in these patients. As mentioned above, endothelial barrier integrity is strongly dependent on cell adhesion complexes formed by VE-cadherin as well as integrins. Both types of complexes are under the control of the cytoskeleton and are regulated by a variety of kinases, which constitute potential drug targets. In addition to the angiopoietin-2/Tie2 axis outlined earlier, several cytoplasmic kinases are known to regulate endothelial adhesion complexes during inflammation. For instance, it was shown recently that the kinase Abl2/Arg is an essential regulator of barrier function in a preclinical model for sepsis [180], and clinical trials with Abl2/Arg inhibitors in ICU-admitted COVID-19 patients are ongoing (NCT04794088) [181].
In summary, the data emerging from several lines of investigation indicate a central role for the endothelium, with interconnected pathways linking inflammation, endothelial injury, and microvascular thrombosis in the pathophysiology of COVID-19. These pathways require further investigation, as does the question of how the virus may initiate the process. Better knowledge of these relationships should reveal multiple targets for new therapies.
Pulmonary vascular endothelialitis and intussusceptive angiogenesis in COVID-19
To study the pathogenesis of COVID-19, autopsy samples from COVID-19 have been compared with influenza-infected and uninfected control lungs. SARS-CoV-2 infection in these COVID-19 patients was confirmed by both antemortem nasal and postmortem PCR testing, in situ hybridization and immunohistochemistry.
Distribution of COVID-19 disease in the lung
Clues to the pathogenesis of severe COVID-19 disease are provided by antemortem chest imaging. SARS-CoV-2 is a respiratory virus with relatively high viral loads shedding into the entire aerodigestive tract [182, 183]. Despite widespread exposure of the airway epithelium [184], chest CT scanning of COVID-19 patients often demonstrates localized disease. The primary anatomic unit of these opacities appears to be secondary lobules. These are polygonal-shaped anatomic compartments in the peripheral lung, supplied by both the pulmonary and bronchial circulation. At autopsy of patients succumbing to COVID-19, gross examination of the lung often reveals cystic changes and evidence of parenchymal blood clots [102]. Also consistent with selective involvement of the lung, rather than global pneumonitis, the mean wet weight of the COVID-19 lungs at autopsy is significantly less than that of influenza-infected lungs, but greater than that of uninfected control lungs [102].
Microscopic evaluation of the autopsy lung consistently demonstrates dilated alveolar ducts potentially contributing to the cystic changes observed in antemortem imaging and autopsy inspection. Air space edema fluid and hyaline membranes, contributing to the ground-glass opacities on CT scan [185, 186], are seen near the dilated airways. The most dramatic microscopic finding, however, is the presence of vascular thrombi. Whereas thrombi have been found in both the pulmonary and systemic circulations, the dominant finding in COVID-19 has been the frequency of microthrombi within alveolar capillaries. Microthrombi in alveolar capillaries are nearly 10-times more frequent in COVID-19 than in severely affected influenza control lungs [102]. The concentration of thrombi in alveolar capillaries, with fewer thrombi in feeding arteries and draining veins, is consistent with a local injury caused by the virus. The complex issue of systemic coagulopathy is addressed elsewhere in this review.
Endothelialitis and endothelial injury
To evaluate the mechanism of vessel injury in COVID-19 patients, corrosion casting and scanning electron microscopy of the autopsy specimens was performed. In corrosion casting the vascular system is perfused with a low viscosity resin that polymerizes within the microvasculature. To visualize the intraluminal polymer, the surrounding tissue is macerated and the remaining cast of the lumen is examined by scanning electron microscopy (SEM). SEM is a high-resolution imaging technique that is sufficiently scalable to characterize the structure of vascular networks as well as individual vessels.
Corrosion casts from COVID-19 patients have demonstrated luminal irregularities and tortuosity consistent with endothelial inflammation and destruction. The endothelial injuries have been confirmed by transmission electron microscopy (TEM). TEM has demonstrated ECs with significant cytoplasmic disruption, dissolution of gap junctions and separation from the basement membrane. Although the morphology of the lung ultrastructure is significantly compromised by autolysis and aldehyde fixation in autopsy specimens, viral particles have been identified within the ECs at various stages of capsid maturation [187]. Immunohistochemistry has confirmed the presence of spike proteins within ECs. In addition, fluorescence in situ hybridization has documented the presence of viral nucleic acids within ECs [188]. Although functional replication of SARS-CoV-2 in ECs remains an open question, the results are consistent with viral infection of ECs in COVID-19.
The targeting of ECs by SARS-CoV-2 provides an explanation for an interesting clinical observation in COVID-19. A subset of patients with COVID-19 lack breathlessness or discomfort in the setting of profound hypoxemia [187,188,191]. In many cases, these patients have dangerously low oxygen levels (arterial oxygen levels less than 60 mmHg). This so-called "happy" or "silent" hypoxia likely reflects the disruption of normal blood flow regulation in the lung. In normal circumstances, blood flow to inefficient regions of the lung is restricted by so-called hypoxic vasoconstriction [192]—a vasoregulatory process that limits blood flow to hypoxic regions of the lung. This regulatory matching of blood flow with ventilation requires the upstream transfer of information mediated by functional ECs [193]. In COVID-19, endothelial injury results in impaired blood flow regulation and the mismatching of lung perfusion and alveolar oxygen. The failure of the lung to limit—and perhaps even augment [194]—blood flow to hypoxic regions results in systemic hypoxemia. In the absence of vasomotor control over pulmonary blood flow, postural measures such as “proning” can have a dramatic effect on improving system oxygenation [195, 196]. We speculate that other organs that are dependent upon perfusion matching, such as the brain, may also be adversely affected by COVID-19 endothelial injury.
Endothelial injury and the immune response
Endothelial injury can be caused by direct cytopathic effects of the virus, the antiviral immune response, or a combination of both. The role of the immune system in the vascular phase of COVID-19 is particularly uncertain given the decline in circulating lymphocyte concentrations, a unique and distinguishing feature of COVID-19. Lymphocyte counts less than 20% of peripheral blood leukocytes have been associated with severe disease and death [197, 198]. It is possible that peripheral blood lymphopenia reflects a reduction in the total body pool of recirculating lymphocytes either from COVID-19-related toxicity or “functional exhaustion” [199]. Another possibility is that the peripheral blood lymphopenia reflects the sequestration of lymphocytes to areas of active inflammation.
Endothelial injury and intussusceptive angiogenesis
Corrosion casting and SEM analysis of COVID-19 lungs has demonstrated significant amounts of “intussusceptive” (nonsprouting) angiogenesis. The process of intussusceptive angiogenesis was initially identified in 1986 [200], although earlier reports described a similar process. The distinctive feature of intussusceptive angiogenesis is the intussusceptive pillar, a cylindrical microstructure that spans the lumen of small vessels and capillaries. The extension of the pillar down the axis of the blood vessel appears to be a mechanism for creating two lumens from a single vessel. This process of vascular duplication has been frequently cited as a mechanism for microvascular network expansion in ischemic tissue because of the minimal requirement for endothelial proliferation [16]. Intussusceptive angiogenesis and angiogenic disorders associated with endothelial dysfunction giving rise to this abnormal vessel phenotype in COVID-19 have been summarized in Figs. 2 and 3.

Autopsy studies of patients dying of COVID-19. a Hematoxylin and eosin histology demonstrated perivascular lymphocytic infiltration (10X, bar = 100um). b Higher resolution (60X) imaging of the alveolar septa demonstrating microthrombus in alveolar capillaries (black arrows). c Scanning electron microscopy of the COVID-19 lung demonstrating preserved architecture with perivascular and interstitial lymphocytes. Intravascular thrombus was visualized in many vessels (white arrows; bar = 200um). d Corrosion casting demonstrating luminal irregularities associated with endothelial injury and endothelialitis. In the affected microcirculation, innumerable intraluminal pillars (circles), seen as small holes in the cast, reflect the process of intussusceptive angiogenesis (bar = 100um)

Intussusceptive angiogenesis: hypothesis for lung vessels modification in COVID-19
Using finite element flow models, it was shown that pillars form in microhemodynamic "dead zones" within the vessel [201, 202]. Pillars form in flow regions with shear stress below 1 dyn/cm2 [202]. Because areas of low shear stress are not always associated with intussusceptive pillars, a permissive role for low shear stress in pillar development was postulated. Regions of low wall shear stress are necessary, but not sufficient, for the development of intraluminal pillars. In addition to low shear stress, EC activation from extravascular and/or intraluminal signals appears to be required for pillar formation [202].
The tissue response to COVID-19 has created a complex, yet distinct transcriptional profile [102]. Using NanoString characterization of gene expression in COVID-19 and influenza lungs, several unique features were identified. Patients with relatively acute clinical courses have lungs with relatively high levels of hypoxia- and ischemia-related gene expression (e.g. HIF1A) at autopsy These observations suggest that the endothelial injury, thrombosis and vascular dysregulation in COVID-19 is associated with tissue ischemia. Perhaps consistent with an acute episode of tissue ischemia, prolonged hospitalization is associated with a decline in ischemia-related gene expression and an increase in parenchymal remodeling gene expression. Patients with a prolonged disease course have commonly demonstrated high expression of genes encoding extracellular matrix proteins, a transcriptional signature associated with injury repair. Importantly, the degree of intussusceptive angiogenesis continues to increase with prolonged hospitalization indicating that intussusceptive angiogenesis is a fundamental mechanism of microvascular expansion and network remodeling.
Host factors and future directions
COVID-19 is associated with distorted microvascular networks and microthrombi in gas exchange regions of the lung. Infection not only disrupts luminal blood flow but also may compromise ventilation/perfusion matching within the lung. The consequence is severe hypoxia and potential tissue ischemia. In normal circumstances, localized tissue ischemia within the lung is compensated by the bronchial circulation. The bronchial vessels arise from the descending aorta to perfuse the proximal airways as well as the fibrous septa of secondary lobules in the lung. Complementing the deoxygenated blood in the pulmonary circulation, the bronchial circulation contributes oxygenated blood from the systemic circulation. The importance of the bronchial circulation in preventing local tissue ischemia is underscored by observations in lung transplantation and airway reconstructive surgery, situations in which the bronchial circulation is disrupted. In these cases, pulmonary emboli or local thrombosis can result in tissue infarction. These observations indicate that respiratory bronchiolitis and vascular endothelialitis alone do not account for the variation in host susceptibility. We speculate that variation in the size of the viral load, viral receptor expression, viral exposure history or even heritable immune reactivity may contribute to severe COVID-19. It is also possible that a proportion of the differences in clinical outcome will be due to variability in the patency and performance of the bronchial circulation. If a component of the severe phase of COVID-19 is due to tissue ischemia and potential necrosis, then patients will certainly benefit from therapeutic measures designed to improve tissue oxygen delivery.
Perfusion defects in COVID-19 ARDS lungs: intensivist’s point of view on endothelium and pulmonary vasculature
There is still an ongoing debate on whether COVID-19 ARDS differs from ARDS of other causes, and if so to which extent. Since such differences could imply to modify the general rules for applying protective invasive mechanical ventilation, specifically regarding the positive end-expiratory positive (PEEP) and tidal volumes (VT) setting targets, and to initiate vascular interventions such as therapeutic anticoagulation and/or thrombolytic treatment. It is therefore of paramount importance to deeply study arguments for such specific features, and to do this during all stages of the disease. One explanation for such specificities is related to an unusual large extent of lung endothelialitis and macro- and particularly micro-thrombosis, affecting lung perfusion, one of the major determinants of gas exchanges, beside alveolar ventilation. In this chapter, we will review some general ARDS findings focusing on lung microcirculation, then expose different means of lung vascular exploration in COVID-19 ARDS and put these results in perspective with (i) biological markers of endothelialitis and thrombosis, (ii) further needed confirmatory studies and (iii) conceivable specific therapeutic options.
General ARDS physiopathology with a focus on lung microcirculation: historical studies, VD/VT prognosis, microcirculation and dead space.
It is of great importance to remember that ARDS is not a specific disease but rather a well-defined syndrome [203, 204] with a great variety of etiologies, including a large part of infectious agents as causative factors. In recent years, a considerable number of clinical studies focused on the optimal invasive mechanical ventilation settings, with the general aim to best recruit collapsed or flooded alveolar territories, while limiting alveolar overdistension in other parts of the lung, therefore achieving so-called “protective mechanical ventilation”. However, relatively less attention was given to lung pulmonary perfusion, despite the fact that studies have documented the importance of pulmonary vascular lesions and vascular obstruction in ARDS more than 30 years ago [205, 206]. In contrast, the importance of ventilation/perfusion mismatches in pulmonary diseases is well recognized. Moreover, the major influence of pulmonary dead space, as a very important physiological parameter, on ARDS vital prognosis is consistently recognized since the publication of a seminal study in 2002 [207]. Of note, a high amount of pulmonary dead space could reflect, beside other factors, severe lung perfusion disturbances and is a hallmark of a worse prognosis in ARDS. In addition to these general ARDS considerations, COVID-19 induces further major vascular alterations.
Pathophysiological signature: mechanics and gas exchange, response to PEEP, recruitment/inflation ratio (R/I)
Profound hypoxemia is a hallmark of COVID-19, accompanied in the early phase of the disease with relatively preserved pulmonary compliance and with decreased CO2 clearance [137]. Such specific features were confirmed by matching with historical ARDS controls, based on oxygenation or compliance parameters [208], even if limitations for such approaches were highlighted [209]. In parallel, the same authors documented a PEEP-response different from non-COVID ARDS, with the hypothesis that PEEP-induced oxygenation improvement in COVID-19 ARDS patients could result more from redistribution of pulmonary blood flow rather than from recruitment of non-aerated alveoli. It should be mentioned that the exact mechanisms of such redistribution are not clearly understood, with the possibility that extensive lung microvascular insults could participate to such patterns [194]. We can propose as a hypothesis that micro- and macro-pulmonary vessel thrombosis and insults could be a key factor at the early phase of COVID-19 ARDS, explaining that many patients exhibit a better oxygenation response to prone positioning than to increasing PEEP. Accordingly, many authors suggested that specific mechanical ventilator settings could be proposed for COVID-19 ARDS. However, others reported, using a recently proposed mechanistic approach, that most of the COVID-19 ARDS exhibited a high recruitment/inflation ratio (R/I), suggesting a ventilatory benefit of rather high PEEP settings [210]. The hypothesis of pulmonary vessel insults as key factor is underlined by COVID-19 induced pulmonary microthrombosis, as impressively shown in some autopsy cases [102]. Altogether, such results could argue for personalization of mechanical ventilation settings, taken into account not only the potential for alveolar recruitment but also the possible effects of high PEEP settings on lung microcirculation. Furthermore, COVID-19 patients often respond to prone positioning, either on invasive or non-invasive ventilation, particularly in the early phase and even if PaO2/FiO2 ratio is above 150 mmHg, upon the current recommendations.
Pathophysiological signature: pulmonary dead space, ventilatory ratio
Data published on very high physiological dead space (VD/VT) values in the early phase of COVID-19 ARDS in 22 consecutive patients [4] are in line with the above-mentioned microthrombosis formation. In parallel, elevated values of circulating ECs (CECs) and of D-dimer were observed. Altogether, these results permitted us to propose the hypothesis of COVID-19-triggered pulmonary microvascular endothelial damage and microthrombosis, even if other factors could contribute to the observed findings, such as added instrumental dead space, PEEP-induced alveolar distension and pulmonary embolism. These results are to be analyzed in parallel with a number of COVID-19 studies reporting high values for another simpler parameter, i.e. the Ventilatory Ratio (VR), proposed as a marker of ventilatory impairment with an established correlation between VD/VT and VR [211]. However, a weaker correlation between VD/VT and VR has been observed in COVID-19 ARDS patients, reinforcing the limits of VR as a marker of non-perfused lung areas, as compared to VD/VT and derived parameters [4, 212]. To determine whether VD/VT shares the same vital prognosis value in COVID-19 ARDS as in non-COVID-19 ARDS will be an important topic of investigation in the future. In light of these data, the authors clearly advocate for a reappraisal of routine dead space measurement at least in COVID-19 ARDS.
Pathophysiological signature: microcirculation studies
Rovas et al. reported sublingual intravital microscopy results obtained in 23 moderate to severe and critical COVID-19 patients, compared to 15 healthy controls [75]. They mainly observed a reduction in vascular density (especially for small capillaries) and in red blood cells velocity. Patients on mechanical ventilation showed severe glycocalyx damage as indicated by higher perfused boundary region. Similar microvascular alterations were observed in severe COVID-19 ARDS patients [74, 213].
Interestingly, Kanoore Edul et al. reported similar decreases in the proportion of perfused vessels and flow velocity in 27 COVID-19 ARDS patients, while they also observed high vascular densities, perhaps in relation with enhanced angiogenesis or capillary recruitment [214]. Finally, Do Espirito Santo reported multiple filling defects suggestive of moving thrombi in 11 of 13 COVID-19 patients requiring invasive mechanical ventilation [215].
Taken together, these results underline the hypothesis of severe microvascular dysfunction and thrombosis in ARDS. In a similar way to non-COVID-19 ARDS patients, such results could reinforce the hypothesis of similar alterations at the lung level, favoring a high number of ventilated alveoli with no or severely diminished perfusion [4, 216, 217].
Based mainly on DECT and CT pulmonary angiogram, Patel et al. documented perfusion defects in 39 invasively mechanically ventilated COVID-19 patients, associated with dilated peripheral vessels ("tree-in-bud" pattern) in 64% of the patients [218]. Importantly, pulmonary macro-embolism was present in only 38% of the patients. Additionally, they documented a hypercoagulable and hypo-fibrinolytic state using thromboelastography. Altogether, these data support the concept of a severe pulmonary thrombotic angiopathy and extend previous findings [219]. Beenen et al. compared dual-energy CT pulmonary angiogram results between 10 mechanically ventilated COVID-19 patients and 10 less severe COVID-19 patients suspected from PE [220]. PE was observed in eight ICU patients and two ward patients. Despite an occurrence of only seven central or segmental PEs, the authors observed much larger areas of perfusion defects (52.5 ± 14.8 and 17.5 ± 7.5% respectively, mean SD) than anticipated, reinforcing the hypothesis of diffuse pulmonary microcirculatory dysfunction associated with microthrombi. They also reported a lower pulmonary blood volume in the more severely ill patients.
Electrical impedance tomography (EIT) studies
Electrical impedance tomography (EIT) is an elegant bedside method to assess regional lung ventilation, but also regional lung perfusion by injection of 5–10% saline solution as contrast agent. Accordingly, it could be a very interesting method for assessing the severe pulmonary ventilation/perfusion imbalances, which could characterize COVID-19 ARDS to some extent. However, spatial resolution is limited now to more central and larger vessels. Unfortunately, only limited data are available combining both ventilation and perfusion assessment in COVID-19 patients. Mauri et al. reported in six out of seven such COVID-19 patients’ large ventilation/perfusion mismatch, with a median (IQR) dead space fraction of 22% [221]. Comparable results were reported in 15 other patients: median (IQR) dead space fraction of 27% [23,24,25,26,27,28,29,30,31,32,33,34,35,36] [222]. Importantly, the percentage of dead space measured by EIT differs from the measures provided by capnography, because it refers to dead space inside the lungs (minimizing the contribution of instrumental and anatomical dead space), with lung aerated volume (and not tidal volume) as the reference value. These two studies therefore strengthen the hypothesis of severe pulmonary microcirculatory dysfunction and thrombosis. Finally, Fakhr et al. also reported lung perfusion EIT data in one mechanically ventilated COVID-19 patient, in which asymmetrical distribution of perfusion led to the diagnosis of PE on CT pulmonary angiogram [223].
Further needed confirmatory studies
Confirming or not that COVID-19 ARDS could be viewed, and to which extent, as a distinct ARDS endotype, named by some authors the "vascular endotype", is a major challenge for the future. To achieve this goal, it will be very important to fully study in COVID-19 ARDS patients pulmonary physiological parameters during the full ICU clinical course in parallel with biological markers (focusing on the coagulation and fibrinolytic systems and on endothelium activation and senescence markers) and microcirculation studies. Pulmonary physiological parameters could be in such future studies derived from capnography measurements (with alveolar dead space as a major study parameter), from EIT studies (with an emphasis on pulmonary perfusion parameters) and from CT studies (including pulmonary blood volume quantification). Another interesting point may be to study to which extent such an ARDS endotype could induce residual clot burden and influence post-ICU pulmonary functional abnormalities described in COVID-19 ARDS, such as a decrease in diffusing capacity of the lung for CO (DLCO) [222,223,226]. Since a decrease in lung capillary blood volume (LCBV) could influence DLCO results, it could be interesting to measure diffusing capacity of the lung for both nitric oxide (NO) and DLCO, permitting to quantify LCBV [227].
Such future studies, in addition to existing knowledge, will contribute to a better description of the COVID-19 ARDS pathophysiology and could allow the identification of a patient's profile in which curative anticoagulant therapy or thrombolytic treatments could be considered. Additionally, specific mechanical ventilator settings may also be suggested.
COVID-19 is an acquired hemopathy
The mammalian innate immune response can detect viral replication and could protect the host against SARS-CoV-2 infection partly through the action of interferons (IFNs) that induce hundreds of genes encoding antiviral effectors. For example, induction of the LY6E (lymphocyte antigen 6 complex, locus E) gene inhibits SARS-CoV-2 entry into cells by impairing spike protein-mediated membrane fusion [228]. Pattern recognition receptors (PRRs) of the innate immune system recognize viral antigen and virus-induced tissue damage, increasing bone marrow hematopoiesis, to release neutrophils and monocytes, and stimulate the production of cytokines and chemokines. Recently, emergency hematopoiesis generating immune-suppressive myeloid cells was detected in the blood of patients with severe COVID-19 [229, 230]. This emergency myelopoiesis generating immature myeloid cells shares several features with acquired hematological malignancies, which will be discussed below.
Homeostatic and regenerative hematopoiesis
Self-renewal and differentiation of hematopoietic stem cells (HSCs) are tightly regulated by intrinsic and extrinsic factors produced by hematopoietic and non-hematopoietic cells of the bone marrow niche. The advent of new technologies such as fate-mapping and in situ labeling have shown that quiescent HSCs with low metabolic activity poorly contribute to blood cell production [231, 232], which mostly involves multipotent progenitors (MPPs) [231, 233]. HSCs differentiate into slowly cycling and metabolically active MPP1 [234] and then into MPPs subsets with preferred lineage potential and rapidly cycling capacity, i.e. myeloid-biased MPP2 and MPP3 and lymphoid-biased MPP4 [231, 233]. MPPs differentiate into common myeloid progenitors, which produce granulocyte/macrophage progenitors (GMPs) and megakaryocyte/erythrocyte progenitors (MEPs) with heterogeneous lineage commitment [235]. High-dimensional flow cytometry indicates that GMPs constitute a pool of cell types, ranging from exclusive neutrophil, monocyte, or dendritic cell progeny to multipotential progenies [236]. Infectious stress induces an adapted hematopoiesis, which involves an increased generation of MPP2 and MPP3 while MPP4 are partly reprogrammed towards the myeloid lineage [237]. GMPs expand and form clusters in the bone marrow niche upon signals delivered by stem cell factor (SCF) and granulocyte colony-stimulating factor (G-CSF) that promote granulocytic differentiation, whereas transforming growth factor (TGF)-β and CXCL4/platelet factor-4 limit dormant HSC activation [238].
Emergency hematopoiesis in response to infection
Myeloid cell response
In response to infectious stress, adaptive immune T and B cells proliferate in an antigen-specific manner while innate immune effector cells, which are rapidly recruited by infected tissues, must be replaced by new cells generated by the hematopoietic stem and progenitor cell (HSPC) pool. This demand-adapted myelopoiesis, which was shown to be associated with acquisition of an innate immune memory, involves a response of HSPCs to inflammatory signals [239]. Importantly, the demand-adapted myeloid output, which usually provides rapid protection against threats, must be transient to prevent HSC exhaustion, restore a balanced production of blood lineages and prevent immune cell-induced toxic effects.
The HSPC response to systemic bacterial and viral infection implicates two non-mutually exclusive mechanisms: (1) a cytokine-mediated mechanism in which pro-inflammatory cytokines, which are secreted locally in the bone marrow, trigger HSC proliferation through their receptors and downstream signaling pathways [238,239,242]; (2) a direct sensing of HSPCs by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) released by tissues after the pathogenic insult. HSPCs recognize PAMPs via pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), which promote their proliferation, differentiation, and migration by triggering transcriptional and epigenetic networks, counterbalancing the loss of mature myeloid cells and generating the innate immune memory [243, 244]. Upon TLR activation, HSPCs produce pro-inflammatory cytokines including IL-6 that enhance proliferation and myeloid differentiation in vivo [245]. Type I IFN may activate dormant HSCs to produce such a response [242]. Long-term HSCs are also able to directly sense PAMPs and exit from dormancy via TLR4 activation [246]. Pharmacological inhibition of this process does not prevent emergency granulopoiesis. By contrast, prolonged TLR4 signaling may contribute to inflammation-associated HSPC dysfunction. Tissue-resident macrophages—as a first line of defense—and non-hematopoietic cells such as ECs and mesenchymal stromal cells (MSCs) also express PRRs and contribute to pathogen sensing [247]. ECs in the bone marrow microenvironment are critical regulators of emergency hematopoiesis through the expression of high amounts of G-CSF in response to lipopolysaccharide (LPS). Finally, long-term innate immune memory or trained immunity involves an IL-1β and GM-CSF-dependent expansion of HSPCs, and the adaptation of monocytes and macrophages to promote the response to a secondary inflammatory insult [248].
Role of pro-inflammatory cytokines
A spectrum of pro-inflammatory cytokines and chemokines regulate HSCs and hematopoiesis. Type I IFNs (including multiple types of IFN-α, IFN-β), are produced mainly by monocytes. Type I IFNs induce an antiproliferative, proapoptotic state in immature hematopoietic cells through the IFN-α/β receptor, composed of IFNAR1 and IFNAR2 subunits. Acute IFN-α exposure leads to HSC proliferation and exhaustion in vivo, while chronic IFN-α exposure rapidly makes these HSCs return to quiescence to ensure survival of the HSC pool [249]. Type II IFN (IFN-γ) is produced by T cells and natural killer (NK) cells, and bind to a receptor consisting of IFNGR1 and IFNGR2 subunits. IFN-γ plays a critical role in cellular immune responses against pathogens via upregulation of MHC class I and II, augmentation of Th1 immune responses, and activation and differentiation of macrophages. Although it has suppressive effects on hematopoietic progenitors, IFN-γ also stimulates HSC proliferation in synergy with IL-3 or IL-6 inducing a transient shift from dormancy to proliferation and differentiation [250].
IL-6 is a multifunctional cytokine important for regulation of hematopoiesis as well as immune responses in acute and chronic inflammation conditions. The IL-6 receptor consists of a ligand-binding IL-6-receptor α chain (IL-6Rα) and signal-transducing subunit β-receptor glycoprotein 130 (gp130) linked to the JAK/STAT pathway. IL-6 is secreted by T cells and macrophages in response to infection or tissue damage resulting in inflammation. In turn, IL-6 stimulates the production of bone marrow neutrophils whereas it is not necessary for maintaining homeostatic blood cell levels. Upon viral infections, IFN-γ promotes the production of IL-6 in the bone marrow MSCs, which increases the number of early MPPs and the accumulation of myeloid cells [251]. IL-3 promotes steady-state hematopoiesis, and contributes to the production of bone marrow monocytes during sepsis, suggesting its involvement in emergency hematopoiesis. Blocking IL-3 release reduces sepsis mortality [252]. The IL-1 cytokine family is critical in the initiation of pro-inflammatory responses to infections by upregulating expression of integrin’s on leukocytes and ECs. Their receptor complex is composed of IL-1RI and IL-1R accessory protein (IL-1RAP). Acute IL-1 signaling in response to infection leads to HSC differentiation biased into the myeloid lineage while chronic exposure promotes uncontrolled HSC division, loss of self-renewal activity, and exhaustion of the HSC pool [253].
G-CSF signals through its homodimer receptor G-CSFR to maintain homeostatic granulopoiesis and induce the production of granulocytes in response to infection. It also mobilizes dormant HSCs to the blood without stimulating their proliferation, while in conditions of bone marrow regeneration, it mediates the proliferation of megakaryocyte-biased HSCs. Macrophage colony-stimulating factor (M-CSF) controls the proliferation, and differentiation of GMPs into monocytes and macrophages by interacting with its receptor M-CSFR, and regulates steady-state myelopoiesis. Both G-CSF and M-CSF serve as major regulators of inflammatory myelopoiesis related to their ability to induce metabolic reprogramming through the mTOR pathway [252,253,256].
An emergency myelopoiesis in severe COVID−19
Immunosuppressive myelopoiesis
In severe COVID-19, the control mechanisms of emergency myelopoiesis may be overflowed (Fig. 4). In addition to tissue damage by the virus, innate immune over−activity contributes to acute lung injury (257). Pneumocyte lysis induced by SARS−CoV−2 provokes the release of large amounts of PAMPs and DAMPs, which may sense HSCs to switch from steady state to emergency hematopoiesis. Our recent observations, corroborated by many others, indicate that, in patients with severe COVID−19, this emergency hematopoiesis is exacerbated and persists (229, 230, 256,257,258,259,262). Similar to bacterial sepsis (263, 264), patients present with neutrophilia, low basophils, decreased plasmacytoid dendritic cells, lymphopenia due to decreased CD4 and CD8 T cells, as well as B cells, with a neutrophil to lymphocyte ratio that is strongly increased (265). CD66b+ granulocytes and immature CD10low CD101+/− neutrophils are present. Like in sepsis, immature neutrophils show increased expression of CD64 and the immune checkpoint molecule programmed death−ligand 1 (PD−L1), a marker of T cell suppression (230) and mature neutrophils express CD177, an anti−inflammatory molecule suggesting dysfunctional neutrophils (266).

Emergency myelopoiesis. Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) released by damaged tissues after the pathogen insult are sensed by tissue-resident macrophages, hematopoietic stem and progenitor cells (HSPC), mesenchymal stroma cells (MSC) and endothelial cells (EC) via the pattern recognition receptors ie Toll-like receptors. These cells release inflammatory cytokines forcing the proliferation and differentiation of HSPC into granulocyte-monocyte progenitors (GMP), which produce neutrophils and monocytes with immunosuppressive phenotype. In severe COVID-19, immature neutrophils and dysfunctional monocytes that resemble myeloid-derived suppressive cells produce high amounts of calprotectin, which in turn amplifies the emergency myelopoiesis. IL-1: interleukin 1, IL-6: interleukin 6, IFN-γ: interferon γ, GM-CSF: granulo-monocyte colony-stimulating factor, G-CSF: granulocyte colony-stimulating factor, M-CSF: monocyte colony-stimulating factor
In severe COVID-19 monocyte blood count is normal but phenotypes are abnormal with accumulation of classical CD14+ CD16− monocytes (MO1) expressing high levels of CD64 (FcγRI) but low levels of HLA−DR, early disappearance of non−classical CD14low CD16+ monocytes (MO3), and abnormal expression of the activation marker CD163 (229, 230). Of note, low HLA−DR expression on monocytes is a surrogate marker of immunosuppression in sepsis or cancer and a high proportion of HLA−DRlow monocytes correlates with death in septic shock (267, 268).
Single−cell transcriptomic analyses of neutrophils and monocytes show unique signatures in severe COVID-19. IFN type I pathways are not activated in either cell type. Immunosuppressive features with activation of reactive oxygen species (ROS) and nitric oxide synthase (NOS) pathways in the monocytes and IL−1, ROS and NF−κB pathways in the neutrophils (229), recapitulating the immunosuppressive functions described in myeloid−derived suppressive cells (MDSCs) often detected in the blood and tissues of patients with cancer, sepsis and severe influenza, are also found in severe COVID−19 (269, 270). Immature neutrophils resemble granulocytic MDSCs expressing S100A8, S1009, S10012 and matrix metallopeptidase 8 (MMP8). Functionally, neutrophils from patients with severe COVID−19 show an impaired oxidative burst response, while their phagocytic capacity is preserved (230). Mononuclear phagocytes and neutrophils predominate in COVID−19 lung infiltrates. By integrating scRNA−seq datasets from bronchoalveolar lavage fluid (BALF) (271) and peripheral blood samples, we observed that alveolar macrophages dramatically decreased in mild and severe patients compared to controls while neutrophils and monocytes/macrophages accumulate. Similar to blood neutrophils, BALF neutrophils have a high expression of S100A8, S100A9, and CXCR4, indicating an immature state. Monocytes/macrophages express IFN−stimulated genes (SIGLEC−1, IFI44, and IFITM3) indicating upregulation of the viral replication and type I IFN signaling pathways. The NOS biosynthetic process and NFκB pathway are upregulated, while HLA−DRA and HLA−DRB1 mRNA expression is low in BALF monocytes/macrophages similar to blood monocytes (229).
Mediators of severity include the overproduction of IL−6 that may participate in the increased production of myeloid cells from HSPCs (272). By contrast, production of IFNα2a or IL−1β is low or decreased compared to controls without SARS−CoV−2 infection (229, 259). Thus, defective or repressed monocyte activation combined with dysregulated granulopoiesis with a transcriptional program related to features of immunothrombosis, may cause a vicious circle of continuous tissue inflammation and ineffective host defense.
A role for megakaryocytes and platelets
Infections and systemic inflammatory responses are associated with life−threatening platelet consumption. The loss of vascular integrity can induce a transient decrease in platelet count whose recovery requires an accelerated maturation of megakaryocytes. In humans, a very small fraction of the long-term (LT)−HSC compartment is constituted by megakaryocyte−biased progenitors and may contribute to steady−state megakaryopoiesis (235, 273). Megakaryocyte priming of the HSC compartment has been robustly demonstrated in mice in which rapid regeneration of the platelet pool involves a megakaryocyte−primed stem−like progenitor, expressing megakaryocyte−specific transcripts that become translated. Increased expression of CD41 at the surface of HSCs is the hallmark of megakaryocyte response to infection in mice. The maturation of megakaryocyte progenitors leads to efficient replenishment of life−threatening platelet depletion during acute inflammation (274, 275). Hematopoietic progenitors have also been detected in healthy lung that may be a primary site for platelet biogenesis, at least in mice (276).
Severe COVID-19 is associated with both thrombocytosis through enhanced generation of platelets, and thrombocytopenia through increased consumption of platelets, e.g. as an effect of extended lung immunothrombosis (277). A high density of alveolar megakaryocytes has been described in some patients suggesting active in situ megakaryopoiesis (278). Local stimulation of hematopoietic cells could contribute to the lung damage through the release of immunomodulatory molecules and the interaction of platelets with granulocytes and monocytes. The presence of megakaryocytes is also reported in the peripheral blood of patients with severe SARS-CoV-2 infection. Megakaryocytes highly express CD41, IFITM3 and IFITM2, suggesting a strong type I IFN response conferring an anti−viral activity to megakaryocytes and platelets (279). Interestingly, TGFB1 gene expression by megakaryocytes is concomitantly decreased. In regenerative conditions, TGFβ and CXCL4/PF4 produced by megakaryocytes located in the vicinity of GMP clusters have been proposed to spatially restrain myelopoiesis in the bone marrow. In severe COVID-19, the lack of TGFβ inhibitory effects could therefore prevent the resolution of emergency myelopoiesis.
Contribution of clonal hematopoiesis and co−morbidities to immunosuppressive myelopoiesis
Uncontrolled emergency myelopoiesis could participate in mediating the poor impact of comorbidities (aging, obesity or cancer) on COVID−19 outcome. Common aging−related changes in hematopoiesis include a skewed differentiation toward the myeloid lineage with lymphoid deficiency and a decreased clonal heterogeneity and regenerative potential, together with a systemic inflammatory status that reduces immune tolerance, an effect known as “inflamm−aging” that predisposes to hematological malignancies and other diseases (280). Mutations might arise through mitochondrial dysfunction, ROS production, increased autophagy, epigenetic changes and DNA damage accumulating in HSCs in an inflammatory bone marrow microenvironment. Mutations affect multiple genes including epigenetic regulators known as driver of HSC decline like TET2, DNMT3A, and ASXL1. A high prevalence of Clonal hematopoiesis of indeterminate potential (CHIP) has been recognized in COVID−19 patients, with a disputed impact on immediate clinical outcome (281, 282). Of note, direct control of important pro−inflammatory proteins is exerted by TET2 through transcription or post−transcriptional regulatory mechanisms. At the chromatin level, TET2 is required to resolve inflammation by specifically repressing IL−6, while TET2 inhibition increases the production of HSPCs; mature myeloid cells and inflammatory cytokines in response to inflammatory stress (283, 284). During bacteria−induced sepsis in mice, Tet2 regulates 5−methylcytosine mRNA oxidation which controls SOCS3 expression and increases myelopoiesis (285). The functional inactivation of TET2 could participate in uncontrolled and deleterious myeloid response.
Obesity may also exaggerate the myelopoietic response to SARS-CoV-2 infection. In mouse models of obesity by high fat diet, excess of pro-inflammatory IL-1 directly acts on the GMP compartment, leading to increased production of monocytes and neutrophils [286]. High fat diet-induced myelopoiesis could also occur via cell-autonomous metabolic regulation of myeloid progenitors.
The S100A8/S100A9 heterodimer a key mediator of emergency hematopoiesis in COVID−19
Severe forms of COVID-19 are associated with a prolonged elevation of many circulating pro−inflammatory soluble mediators like IL−6 in the peripheral blood. Next to increased presence of these molecules, we detect a massive release of the heterodimer S100A8/S100A9 (calprotectin) whose genes are also the most highly expressed genes in innate immune cells that accumulate in the blood of severe COVID-19 patients (229). Elevation of peripheral blood calprotectin is confirmed to be associated with severe outcome in SARS−CoV−2−infected patients by several independent groups (285,286,289).
S100A8 and S100A9 alarmins are Ca2+ binding proteins representing ~45% of the cytoplasmic protein pool in neutrophils. Low levels of S100A8/A9 are constitutively expressed in monocytes but intensely upregulated in inflammatory settings (290). S100A8/A9 can signal through TLR4, RAGE (receptor for advanced glycation end products) and CD33, depending on the target cell and the context.
S100A8/A9 plays an important role in protecting the body of pathogenic infection, i.e. in controlling neutrophil migration during inflammation [290]. An interesting example is the prominent role for S100A8/9 in the imprinting of a functional specialization of monocytes to allow colonization by microbes within newborns [291].
If proper levels of S100 proteins contribute to defense capabilities and immunity homeostasis, excessive amounts of S100A8/A9 magnify the inflammatory response and accelerate cytokine release by neutrophils and monocytes, generating a feed−forward vicious cycle. Alarmins promote an overproduction of myeloid cells in various disease settings, including autoimmune diseases, myocardial infarction, and diabetes (290,291,294). In our non−infected, control cohort, we detected elevated plasma levels of calprotectin in patients with these comorbidities. Thus, people who are most at risk of severe COVID−19 demonstrate abnormally high basal levels of circulating alarmins. Nevertheless, these levels always remain significantly lower than those measured in severe COVID−19 patients (229).
Further supporting a role for calprotectin in a toxic feedback loop involving HSPCs, alarmins produced by bone marrow macrophages and erythroblasts are involved in the pathophysiology and severity of myelodysplastic syndromes (MDS) by promoting the generation of dysplastic and immunosuppressive myeloid cells while inhibiting erythropoiesis through paracrine and autocrine mechanisms [295]. MDS HSPCs overexpress alarmins that trigger pyroptosis of these cells [296]. In addition, transgenic S100A9 mice display bone marrow accumulation of MDSCs accompanied by development of progressive MDS-like disease [297]. MDSCs overexpress alarmins, which mediate premature death of HSPCs. Interestingly, rHuS100A9 can induce the expression of programmed cell death protein 1 (PD-1) on HSPCs [298]. Therefore, alarmins could be involved in the induction of immune checkpoint and impairment of immune responses through T-cell exhaustion and apoptosis. Finally, in patients with a myeloproliferative neoplasm, stroma cells produce calprotectin that triggers myelofibrosis [299]. The contribution of calprotectin to the initiation and support of stress hematopoiesis and immunosuppression in the COVID-19 needs further investigation.
Endothelial progenitor cells as a source of immature cells in the vascular remodeling process observed in COVID-19
Numerous cellular and genetic studies, in particular during embryogenesis, have proposed a developmental relationship between hematopoietic and EC lineages. The hemangioblast has been proposed as a common stem cell for both lineages. More recently, human bone marrow very small embryonic-like stem cells (VSELs) have been described to give rise to both the endothelial and hematopoietic lineage [300]. Circulating endothelial progenitors cells (EPCs) have been widely described in the past 20 years as good biomarkers of cardiovascular, thrombotic and infectious diseases, as well as in cancer. Mancuso et al. reported an increase of CD146 + EPCs in COVID-19 patients compared with healthy controls [301]. This increase was observed in patients with mild symptoms and not further augmented in patients with severe symptoms. In patients who recovered, EPCs decreased, but were still in a significantly higher range than normal controls. The authors also found a positive correlation between the copy number of SARS-CoV-2 RNA in the cellular fraction and apoptotic EPCs per milliliter in severe COVID-19 patients. Circulating EPC phenotypes have been widely discussed and the relevance of each subtype is always a matter of debate. Therefore circulating EPCs are also referred to as CD34+KDR+ cells. Guerin et al. quantified the pool of circulating CD34+KDR+ cells in ten patients with different COVID-19 severity [302]. Intriguingly, COVID-19 at critical stage was associated with a redistribution of circulating CD34+ and CD19+ sub-populations in peripheral blood. KDR was only expressed by CD19+ B-lymphocytes and CD14+ monocytes subpopulations in circulation. Furthermore, a significant mobilization of CD34+c-Kit+KDR− and CD19+KDR+ cells was observed between moderate and critical COVID-19 patients. Mobilization of these different vasculogenic subtypes could be a consequence of hypoxia and/or inflammation associated with COVID-19. However, EPCs increase in severe COVID-19 could participate newly formed vessels and intussusceptive angiogenesis observed in the lungs as proposed in Fig. 3.
Immune reaction, vaccination and thrombosis
Going largely unnoticed, a major trend has been reported initially in China, then in Europe and finally in the USA: the antibodies raised after being infected by the SARS-CoV-2 may not be protective against clinical symptoms, including severe cardiovascular manifestations. To date, this pattern seems to be associated with natural antibodies (found after infection), not with the antibodies raised after vaccination.
First reports from China
Since the publication of the first report form China about patients infected in January and February 2020 the same trend has been found: while a strong production of anti-SARS-CoV-2 antibodies was measured in infected patients, this immune response to infection was not associated with protection against severe manifestation of the disease, including its cardiovascular manifestations. In fact, the opposite has been reported most of the time. One of the first reports, by Qu et al. [303] profiled the serological responses to SARS-CoV-2 nucleocapsid (N) protein and spike (S) glycoprotein in 394 COVID-19 patients admitted to The Third People’s Hospital of Shenzhen between January 11th and February 10th, 2020. The majority of these patients developed robust antibody responses, with a stronger antibody response in critically ill patients. Both the mean IgG and IgM response of this critical group were significantly stronger than that of the non-critical group within four weeks after illness onset (97.85 AU/ml vs 65.76 AU/ml p = 0.0001 and 41.56 AU/ml vs 21.38 AU/ml, respectively). Another early study [304] described the acute antibody responses to SARS-CoV-2 in 285 patients with COVID-19 and found that within 19 days after symptom onset, 100% of patients tested positive for antiviral IgG. Both IgG and IgM titers plateaued within 6 days after seroconversion. Interestingly, the IgG response 8–14 days after the onset of symptoms was significantly higher in the severe patients than in the non-severe patients (p = 0.001). Zaho et al. [305] conducted a study with 173 patients with COVID-19 admitted to the hospital between January 11th and February 9th, 2020. All enrolled cases were confirmed to be infected with SARS-CoV-2 by use of real-time RT-PCR on samples from the respiratory tract. The quantitative data revealed significantly higher antibody titers (P = 0.004) in patients from the critical compared to patients in the non-critical groups. This result was confirmed by a multivariate longitudinal analysis showing that age (β = 0.139, P < 0.001) and antibody titer (β = 0.336, P = 0.006) were independent factors strongly associated with severity of illness.
Long et al. [306] from the Wenzhou District Centers for Disease Control and Prevention (CDC) conducted extensive RT–PCR screening for 2088 close contacts under quarantine. Among the patients infected with SARS-CoV-2, 60 had no symptoms in the preceding 14 days according to local CDC records, and were transferred to a government-designated hospital for centralized isolation. On admission, 17 individuals were excluded for mild or atypical symptoms and six individuals who developed symptoms four to 17 days after admission were excluded. Finally, 37 asymptomatic cases, defined as individuals with a positive RT-PCR but without any relevant clinical symptoms in the preceding 14 days and during hospitalization, were included in this study. Overall, 178 patients with confirmed SARS-CoV-2 infections were identified in the Wenzhou District before April 10th, 2020, and in this study, the proportion of patients with asymptomatic infections was 20.8% (37/178). Interestingly, the virus-specific IgG levels in the asymptomatic group (median S/CO, 3.4; IQR, 1.6–10.7) were significantly lower (p = 0.005) relative to the symptomatic group (median S/CO, 20.5; IQR, 5.8–38.2) confirming earlier reports from China that the ability to produce antibodies against SARS-CoV-2 was not correlated with a better clinical outcome.
Reports from other countries
In a recent paper, McAndrews et al. [307] studied sera from patients infected in February by SARS-CoV-2. The sera were obtained from the Memorial Hermann Hospital- Texas Medical Center (n = 20). Quantitative detection of IgG antibodies to the receptor-binding domain (RBD) or other regions in the spike glycoprotein in patient samples was not always associated with faster recovery, compared to patients with borderline antibody responses to the RBD. Phipps et al. [308] studied a total of 967 subjects from the University of Texas Southwestern Medical Center, Dallas, Texas who were tested for IgG antibodies reactive to SARS-CoV-2. One-hundred seventy-three cases of confirmed or suspected SARS-CoV-2 were tested for IgG. A subgroup of 37 SARS-CoV-2 PCR-positive cases was tested for nucleocapsid-specific IgM antibody and antibody levels were compared between disease severity groups. No association was observed between mild and severe disease course with respect to IgG and IgM levels, with index values of IgG and IgM antibodies not predicting disease severity in this patient population.
Lynch et al. [309] analyzed the sera (n = 533) from patients from the Zuckerberg San Francisco General Hospital with RT-PCR confirmed COVID-19 (n = 94 with acute infections and n = 59 convalescent patients) using a high-throughput quantitative IgM and IgG assay that detects antibodies to the spike protein RBD and the nucleocapsid protein. Individual and serial samples covered the time of initial diagnosis, during the disease course, and following recovery. They then evaluated antibody kinetics and the correlation between magnitude of the response and disease severity. Compared to those with milder disease, peak antibody levels were significantly higher for patients admitted to the intensive care unit for all time intervals between 6 and 20 days for IgM, and all intervals after 5 days for IgG. Overall, IgM and IgG responses were significantly higher in patients with severe compared with mild disease. Hu et al. [310] found among patients from Georgia with COVID-19 that IgM levels increased early after symptom onset for those with mild and severe disease, but that IgG levels increased early only in those with severe disease. A similar pattern was observed in a separate serosurveillance cohort. Young et al. [311] evaluated the immune response characteristics and the association with clinical severity in a prospective observational cohort study of 100 patients from Singapore with PCR-confirmed SARS-CoV-2 infection (mean age 46 years, 56% male, 38% with comorbidities). Seroconversion occurred at a median of 12.5 days (IQR 9–18) for IgM and 15.0 days (IQR 12–20) for IgG; 54/62 patients (87.1%) sampled at day 14 or later seroconverted. As with the other studies reported here, severe infections were associated with earlier seroconversion and higher peak IgM and IgG levels and the authors concluded that a stronger antibody response was associated with disease severity.
The pattern is consistent irrespective of the nature of the studied antibodies
Amrun et al. [312] reported that the lack of clinical protection associated with the natural production of anti-SARS-CoV-2 antibodies is independent of the epitopes that these antibody bind to. They tested binding to four immunodominant epitopes, S14P5, S20P2, S21P2 and N4P5 on the viral S and nucleocapsid (N) proteins. IgG responses to these epitopes were strong, with N4P5 achieving the highest level of specificity (100%) and sensitivity (> 96%) against SARS-CoV-2. Comparing the binding of antibody to these epitopes, the magnitude of IgG responses to S14P5, S21P2 and N4P5 were strongly associated with disease severity.
A counter argument against the lack of protection provided by natural antibodies against SARS-CoV-2 could be the missing information about their functional abilities to neutralize the virus. In other words, were these antibodies neutralizing or not? This question is of importance as neutralizing antibodies are essential for protection and are thus the main objective to achieve in vaccine development. However, even when measuring neutralizing antibodies, the same pattern of increased titers in more severely ill patients was reported by Ren et al. [313]. In their study, the neutralizing antibodies and antibodies in the longitudinal plasma samples from the LOTUS China trial targeting N, S and RBD associated antigens, were measured by a microneutralization assay and ELISA. Viral load was determined by real-time RT-PCR. A total of 576 plasma and 576 throat swabs was collected from 191 COVID-19 patients. Overall, for IgM and IgG antibody titers against N, S, or RBD or neutralization functionality, it was found that the highest reactivity was detected in the most severe COVID-19 patients and did not clearly correlate with clinical outcomes.
In another study, Wang et al. [314] and Casadevall et al. [315] reported on the kinetics of viral load and antibody responses of 23 individuals with COVID-19 with mild and severe disease. These authors found that high levels of neutralizing antibodies were induced after about 10 days post-onset in both severe and mild patients. They also found that the titer of the neutralizing antibodies was higher in the severe group.
In another approach, Luo et al. [316] investigated the kinetics of IgG avidity maturation during SARS-CoV-2 infection. The IgG avidity assay used a novel label-free immunoassay technology. It was found that there was a strong correlation between IgG avidity and days since symptom onset, and peak readings were significantly higher in severe than mild disease cases.
Thus, an unexpected tendency emerges when looking at the reports trying to correlate clinical outcomes and antibody production in patients infected withSARS-CoV-2: infection-induced antibodies may not be as protective as expected. This lack of protective correlation may be one of the reasons behind the disappointing therapeutic efficacy of convalescent plasma therapy. Indeed, after a promising first observational study [317] and good results in patients with acquired immunodeficiency [318, 319], several double blind, placebo controlled clinical trials failed to show any efficacy for this approach [320, 321]. In this context, the recent success of the mRNA- and viral-vector based vaccines against SARS-CoV-2 are of particular interest and raise several hypotheses: (i) antibodies against SARS-CoV-2 are more efficient as a prophylactic treatment than a therapeutic one, (ii) the antibodies generated by the vaccines are different from the naturally occurring ones and/or (iii) other components of the immune system are triggered by the vaccines. In this context it is important to realize that efficiently neutralizing antibodies are directed against vital epitopes, mainly in the S protein, to target cell entry of the virus. Some of these epitopes can be protected by the virus through conformational changes in the RBD domain. Therefore, targeted vaccination approaches [322, 323] against these vital epitopes, predicted to be preferably present in future SARS strains through evolutionary conservation analysis, will be of major importance in the future.
Reports from thrombotic events after COVID-19 vaccination
Following some cases of thrombosis after vaccination, Oxford-AstraZeneca vaccine (AZD1222) was suspended by some European countries. The European Medicines Agency (EMA) concluded that the benefits of the vaccine in combating COVID-19 outbreak continue to outweigh the risk of side effects. A descriptive analysis of the anti-SARS-CoV-2 vaccination thrombotic risk reported to the World Health Organization (WHO) Global Database for Individual Case Safety Reports (VigiBase) allowed to describe that thrombotic events, including cerebral thrombophlebitis, might occur in association with all three vaccines studied (Pfizer/BioNTech, Moderna and Oxford-Astra-Zeneca) [324]. Moreover, several cases of heparin-induced thrombocytopenia-like syndrome have been described [325, 326]. These two first papers proposed to name this phenomenon Vaccine-Induced Immune Thrombotic Thrombocytopenia (VITT).
All in all, a benefit of the vaccine is a non-discussion point considering COVID-19 outbreak epidemiology. However, there is an urgent need for a prospective evaluation of coagulopathy and thrombotic events to fathom rare but serious side effects after COVID-19 vaccination and better characterize VITT and other thrombotic events after COVID-19 vaccination.
Conclusions and perspectives
Vascular endothelium provides a crucial interface between the blood compartment and tissues and maintains homeostasis. The COVID-19 outbreak has emphasized the interrelation between the pathophysiology of respiratory disease and vascular disorders, and different pathological pathways involved in this disease all have endothelial dysfunction as a hallmark. Direct infection of endothelial cells with SARS-CoV-2 is a matter of debate and probably other conditions such as systemic inflammation or deregulated immunology are a center point of observed endotheliopathy. Large biomarker studies and tissular exploration after autopsies allowed confirming early predictive value of endothelial dysfunction, deregulation of angiogenesis, calprotectin associated to stress hematopoiesis and immunosuppression. Even if we have learned a lot over the past year about COVID-19 pathophysiology, the treatment area is yet to open and we could expect treatments with more accuracy on endotheliopathy, angiogenesis and emergency myelopoiesis to be discovered.
References
Debuc B, Smadja DM (2021) Is COVID-19 a new hematologic disease? Stem Cell Rev Rep 17(1):4–8
Wu Z, McGoogan JM (2020) Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center For Disease Control And Prevention. JAMA 323(13):1239–1242
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382(18):1708–1720
Diehl JL, Peron N, Chocron R, Debuc B, Guerot E, Hauw-Berlemont C et al (2020) Respiratory mechanics and gas exchanges in the early course of COVID-19 ARDS: a hypothesis-generating study. Ann Intensive Care 10(1):95
Karagiannidis C, Mostert C, Hentschker C, Voshaar T, Malzahn J, Schillinger G et al (2020) Case characteristics, resource use, and outcomes of 10 021 patients with COVID-19 admitted to 920 German hospitals: an observational study. Lancet Respir Med 8(9):853–862
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS et al (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26(7):1017–1032
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood 135(23):2033–2040
Helms J, Severac F, Merdji H, Angles-Cano E, Meziani F (2020) Prothrombotic phenotype in COVID-19 severe patients. Intensive Care Med 46(7):1502–1503
Shah A, Donovan K, McHugh A, Pandey M, Aaron L, Bradbury CA et al (2020) Thrombotic and haemorrhagic complications in critically ill patients with COVID-19: a multicentre observational study. Crit Care 24(1):561
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM et al (2020) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 191:148–150
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145–147
Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F et al (2020) Pulmonary embolism in patients with COVID-19: awareness of an increased prevalence. Circulation 142(2):184–186
Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P et al (2020) Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7(8):e575–e582
Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H et al (2020) Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet 396(10247):320–332
Mentzer SJ, Konerding MA (2014) Intussusceptive angiogenesis: expansion and remodeling of microvascular networks. Angiogenesis 17(3):499–509
Helms J, Severac F, Merdji H, Angles-Cano E, Meziani F (2020) Prothrombotic phenotype in COVID-19 severe patients. Intensive Care Med 46:1502–1505
Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN (2020) Cardiovascular disease, drug therapy, and mortality in Covid-19. N Engl J Med 382(25):e102
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W et al (2020) Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med. 382(17):e38
Jonigk D, Markl B, Helms J (2021) COVID-19: what the clinician should know about post-mortem findings. Intensive Care Med 47(1):86–89
Planquette B, Le Berre A, Khider L, Yannoutsos A, Gendron N, de Torcy M et al (2021) Prevalence and characteristics of pulmonary embolism in 1042 COVID-19 patients with respiratory symptoms: a nested case-control study. Thromb Res 197:94–99
Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A et al (2020) Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med 173(4):268–277
Jimenez D, Garcia-Sanchez A, Rali P, Muriel A, Bikdeli B, Ruiz-Artacho P et al (2020) Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest 150:1182–1196
Suh YJ, Hong H, Ohana M, Bompard F, Revel MP, Valle C et al (2020) Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology. https://doi.org/10.1148/radiol.2020203557
Zhang R, Ni L, Di X, Wang X, Ma B, Niu S et al (2020) Prevalence of venous thromboembolic events in novel coronavirus disease-2019 patients: systematic review and meta-analysis. J Vasc Surg Venous Lymphatic Disorders. https://doi.org/10.1016/j.jvsv.2020.11.023
Malas MB, Naazie IN, Elsayed N, Mathlouthi A, Marmor R, Clary B (2020) Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: A systematic review and meta-analysis. EClinicalMedicine. 29:100639
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418
Delabranche X, Quenot JP, Lavigne T, Mercier E, Francois B, Severac F et al (2016) Early detection of disseminated intravascular coagulation during septic shock: a multicenter prospective study. Crit Care Med 44(10):e930–e939
Iba T, Di Nisio M, Thachil J, Wada H, Asakura H, Sato K et al (2016) Revision of the Japanese Association for Acute Medicine (JAAM) disseminated intravascular coagulation (DIC) diagnostic criteria using antithrombin activity. Crit Care 20:287
Taylor FB Jr, Toh CH, Hoots WK, Wada H, Levi M (2001) Scientific Subcommittee on Disseminated Intravascular Coagulation of the International Society on T, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 86(5):1327–1330
Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N et al (2013) A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med 41(9):2069–2079
Helms J, Severac F, Merdji H, Clere-Jehl R, Francois B, Mercier E et al (2020) Performances of disseminated intravascular coagulation scoring systems in septic shock patients. Ann Intensive Care 10(1):92
Khinda J, Janjua NZ, Cheng S, van den Heuvel ER, Bhatti P, Darvishian M (2021) Association between markers of immune response at hospital admission and COVID-19 disease severity and mortality: a meta-analysis and meta-regression. J Med Virol 93(2):1078–1098
Goudot G, Chocron R, Augy JL, Gendron N, Khider L, Debuc B et al (2020) Predictive factor for COVID-19 worsening: insights for high-sensitivity troponin and D-dimer and correlation with right ventricular afterload. Front Med (Lausanne). 7:586307
Khider L, Gendron N, Goudot G, Chocron R, Hauw-Berlemont C, Cheng C et al (2020) Curative anticoagulation prevents endothelial lesion in COVID-19 patients. J Thromb Haemost 18(9):2391–2399
Chocron R, Duceau B, Gendron N, Ezzouhairi N, Khider L, Trimaille A et al (2021) D-dimer at hospital admission for COVID-19 are associated with in-hospital mortality, independent of venous thromboembolism: Insights from a French multicenter cohort study. Arch Cardiovasc Dis. https://doi.org/10.1016/j.acvd.2021.02.003
Susen S, Tacquard CA, Godon A, Mansour A, Garrigue D, Nguyen P et al (2020) Prevention of thrombotic risk in hospitalized patients with COVID-19 and hemostasis monitoring. Crit Care 24(1):364
Delabranche X, Helms J, Meziani F (2017) Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care 7(1):117
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 46(4):586–590
Remmelink M, De Mendonca R, D’Haene N, De Clercq S, Verocq C, Lebrun L et al (2020) Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit Care 24(1):495
Grimmer B, Kuebler WM (2017) The endothelium in hypoxic pulmonary vasoconstriction. J Appl Physiol (1985) 123(6):1635–1646
Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ (1999) Hypoxia/Hypoxemia-Induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler Thromb Vasc Biol 19(9):2029–2035
Gupta N, Zhao YY, Evans CE (2019) The stimulation of thrombosis by hypoxia. Thromb Res 181:77–83
Gendron N, Dragon-Durey MA, Chocron R, Darnige L, Jourdi G, Philippe A et al (2021) Lupus anticoagulant single positivity at acute phase is not associated with venous thromboembolism or in-hospital mortality in COVID-19. Arthritis Rheumatol. https://doi.org/10.1002/art.41777
Oudemans-van Straaten HM (2015) Hemostasis and thrombosis in continuous renal replacement treatment. Semin Thromb Hemost 41(1):91–98
Ranucci M, Ballotta A, Di Dedda U, Bayshnikova E, Dei Poli M, Resta M et al (2020) The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost 18(7):1747–1751
Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L et al (2020) Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost 18(9):2215–2219
Maatman TK, Jalali F, Feizpour C, Douglas A 2nd, McGuire SP, Kinnaman G et al (2020) Routine venous thromboembolism prophylaxis may be inadequate in the hypercoagulable state of severe coronavirus disease 2019. Crit Care Med 48(9):e783–e790
Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V et al (2020) Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost 18(7):1738–1742
Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E et al (2020) COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost 120(6):998–1000
Weiss E, Roux O, Moyer JD, Paugam-Burtz C, Boudaoud L, Ajzenberg N et al (2020) Fibrinolysis resistance: a potential mechanism underlying COVID-19 coagulopathy. Thromb Haemost 120(9):1343–1345
Hattori N, Sisson TH, Xu Y, Desai TJ, Simon RH (1999) Participation of urokinase-type plasminogen activator receptor in the clearance of fibrin from the lung. Am J Physiol 277(3):L573–L579
Bajou K, Herkenne S, Thijssen VL, D'Amico S, Nguyen N-Q-N, Bouché A, Tabruyn S, Srahna M, Carabin J-Y, Nivelles O, Paques C, Cornelissen I, Lion M, Noel A, Gils A, Vinckier S, Declerck PJ, Griffioen AW, Dewerchin M, Martial JA, Carmeliet P, Struman I (2014) PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nature Medicine 20(7):741–747. https://doi.org/10.1038/nm.3552
Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS (2002) Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential. Circulation 106(18):2372–2378
Flevaris P, Vaughan D (2017) The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost 43(02):169–177
Yang L-L, Yang T (2020) Pulmonary rehabilitation for patients with coronavirus disease 2019 (COVID-19). Chronic Dis Transl Med 6(2):79–86
Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S et al (2020) Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J Am Coll Surg. https://doi.org/10.1016/j.jamcollsurg.2020.05.007
Gue YX, Gorog DA (2017) Importance of endogenous fibrinolysis in platelet thrombus formation. Int J Mol Sci. https://doi.org/10.3390/ijms18091850
Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ et al (1989) Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest 84(2):695–705
Undas A, Natorska J (2019) Improving fibrinolysis in venous thromboembolism: impact of fibrin structure. Expert Rev Hematol 12(8):597–607
Kruse JM, Magomedov A, Kurreck A, Münch FH, Koerner R, Kamhieh-Milz J et al (2020) Thromboembolic complications in critically ill COVID-19 patients are associated with impaired fibrinolysis. Crit Care 24(1):676
Thachil J, Juffermans NP, Ranucci M, Connors JM, Warkentin TE, Ortel TL et al (2020) ISTH DIC subcommittee communication on anticoagulation in COVID-19. J Thromb Haemost 18(9):2138–2144
Jonmarker S, Hollenberg J, Dahlberg M, Stackelberg O, Litorell J, Everhov AH et al (2020) Dosing of thromboprophylaxis and mortality in critically ill COVID-19 patients. Crit Care 24(1):653
Helms J, Severac F, Merdji H, Schenck M, Clere-Jehl R, Baldacini M et al (2021) Higher anticoagulation targets and risk of thrombotic events in severe COVID-19 patients: bi-center cohort study. Ann Intensive Care 11(1):14
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 46(6):1089–1098
Paranjpe I, Fuster V, Lala A, Russak AJ, Glicksberg BS, Levin MA et al (2020) Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J Am Coll Cardiol 76(1):122–124
Investigators I, Sadeghipour P, Talasaz AH, Rashidi F, Sharif-Kashani B, Beigmohammadi MT et al (2021) Effect of intermediate-dose vs standard-dose prophylactic anticoagulation on thrombotic events, extracorporeal membrane oxygenation treatment, or mortality among patients with COVID-19 admitted to the intensive care unit: the INSPIRATION randomized clinical trial. JAMA. 325(16):1620–1630
Griffioen AW, Molema G (2000) Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev 52(2):237–268
Angus DC, van der Poll T (2013) Severe sepsis and septic shock. N Engl J Med 369(9):840–851
Weinbaum S, Cancel LM, Fu BM, Tarbell JM (2020) The glycocalyx and its role in vascular physiology and vascular related diseases. Cardiovasc Eng Technol. 12:37–71
Uchimido R, Schmidt EP, Shapiro NI (2019) The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit Care 23(1):16
Song JW, Zullo JA, Liveris D, Dragovich M, Zhang XF, Goligorsky MS (2017) Therapeutic restoration of endothelial glycocalyx in sepsis. J Pharmacol Exp Ther 361(1):115–121
Carsetti A, Damiani E, Casarotta E, Scorcella C, Domizi R, Montomoli J et al (2020) Sublingual microcirculation in patients with SARS-CoV-2 Undergoing Veno-Venous Extracorporeal Membrane Oxygenation. Microvasc Res 132:104064
Damiani E, Carsetti A, Casarotta E, Scorcella C, Domizi R, Adrario E et al (2020) Microvascular alterations in patients with SARS-COV-2 severe pneumonia. Ann Intensive Care 10(1):60
Rovas A, Osiaevi I, Buscher K, Sackarnd J, Tepasse PR, Fobker M et al (2020) Microvascular dysfunction in COVID-19: the MYSTIC study. Angiogenesis. 24:145–157
Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS (2020) Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med 8(7):681–686
Drost CC, Rovas A, Kusche-Vihrog K, Van Slyke P, Kim H, Hoang VC et al (2019) Tie2 activation promotes protection and reconstitution of the endothelial glycocalyx in human sepsis. Thromb Haemost 119(11):1827–1838
Rovas A, Seidel LM, Vink H, Pohlkotter T, Pavenstadt H, Ertmer C et al (2019) Association of sublingual microcirculation parameters and endothelial glycocalyx dimensions in resuscitated sepsis. Crit Care 23(1):260
Lee DH, Dane MJ, van den Berg BM, Boels MG, van Teeffelen JW, de Mutsert R et al (2014) Deeper penetration of erythrocytes into the endothelial glycocalyx is associated with impaired microvascular perfusion. PLoS ONE 9(5):e96477
Stahl K, Gronski PA, Kiyan Y, Seeliger B, Bertram A, Pape T et al (2020) Injury to the endothelial glycocalyx in critically Ill patients with COVID-19. Am J Respir Crit Care Med 202(8):1178–1181
Fraser DD, Patterson EK, Slessarev M, Gill SE, Martin C, Daley M et al (2020) Endothelial Injury and glycocalyx degradation in critically Ill coronavirus disease 2019 patients: implications for microvascular platelet aggregation. Crit Care Explor. 2(9):e0194
Potje SR, Costa TJ, Fraga-Silva TFC, Martins RB, Benatti MN, Almado CEL et al (2021) Heparin prevents in vitro glycocalyx shedding induced by plasma from COVID-19 patients. Life Sci. 276:119376
Buijsers B, Yanginlar C, de Nooijer A, Grondman I, Maciej-Hulme ML, Jonkman I et al (2020) Increased Plasma Heparanase Activity in COVID-19 Patients. Front Immunol. 11:575047
Tandon R, Sharp JS, Zhang F, Pomin VH, Ashpole NM, Mitra D et al (2021) Effective inhibition of SARS-CoV-2 entry by heparin and enoxaparin derivatives. J Virol. https://doi.org/10.1128/JVI.01987-20
Gowd V, Gurukar A, Chilkunda ND (2016) Glycosaminoglycan remodeling during diabetes and the role of dietary factors in their modulation. World J Diabetes 7(4):67–73
Kim SY, Jin W, Sood A, Montgomery DW, Grant OC, Fuster MM et al (2020) Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antiviral Res. 181:104873
Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD et al (2000) Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 6(4):460–463
Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V et al (1996) Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 87(7):1161–1169
Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G et al (2015) Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat Commun 6:5962
Hakanpaa L, Kiss EA, Jacquemet G, Miinalainen I, Lerche M, Guzman C et al (2018) Targeting beta1-integrin inhibits vascular leakage in endotoxemia. Proc Natl Acad Sci USA 115(28):E6467–E6476
Kumpers P, Lukasz A, David S, Horn R, Hafer C, Faulhaber-Walter R et al (2008) Excess circulating angiopoietin-2 is a strong predictor of mortality in critically ill medical patients. Crit Care 12(6):R147
Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA et al (2006) Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS medicine. 3(3):e46
Sack KD, Kellum JA, Parikh SM (2020) The angiopoietin-Tie2 pathway in critical illness. Crit Care Clin 36(2):201–216
Smadja DM, Guerin CL, Chocron R, Yatim N, Boussier J, Gendron N et al (2020) Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 23(4):611–620
Vassiliou AG, Keskinidou C, Jahaj E, Gallos P, Dimopoulou I, Kotanidou A et al (2021) ICU admission levels of endothelial biomarkers as predictors of mortality in critically Ill COVID-19 patients. Cells 10(1):186
Kumpers P, Lukasz A (2018) The curse of angiopoietin-2 in ARDS: on stranger TI(E)des. Crit Care 22(1):44
Bermejo-Martin JF, Gonzalez-Rivera M, Almansa R, Micheloud D, Tedim AP, Dominguez-Gil M et al (2020) Viral RNA load in plasma is associated with critical illness and a dysregulated host response in COVID-19. Crit Care 24(1):691
Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J et al (2020) Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ 10(4):2045894020966547
Smadja DM, Philippe A, Bory O, Gendron N, Beauvais A, Gruest M et al (2021) Placental growth factor level in plasma predicts COVID-19 severity and in-hospital mortality. J Thromb Haemost. https://doi.org/10.1111/jth.15339
Byzova TV (2016) “Fishing” out the real VEGFs. Blood 128(19):2283–2284
Kong Y, Han J, Wu X, Zeng H, Liu J, Zhang H (2020) VEGF-D: a novel biomarker for detection of COVID-19 progression. Crit Care 24(1):373
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383(2):120–128
Ramjiawan RR, Griffioen AW, Duda DG (2017) Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 20(2):185–204
Sela S, Natanson-Yaron S, Zcharia E, Vlodavsky I, Yagel S, Keshet E (2011) Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ Res 108(9):1063–1070
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S et al (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111(5):649–658
Dupont V, Kanagaratnam L, Goury A, Poitevin G, Bard M, Julien G et al (2020) Excess soluble fms-like tyrosine kinase 1 correlates with endothelial dysfunction and organ failure in critically ill COVID-19 patients. Clin Infect Dis. 72:1834–1837
Giardini V, Carrer A, Casati M, Contro E, Vergani P, Gambacorti-Passerini C (2020) Increased sFLT-1/PlGF ratio in COVID-19: a novel link to angiotensin II-mediated endothelial dysfunction. Am J Hematol 95(8):E188–E191
Negro A, Fama A, Penna D, Belloni L, Zerbini A, Giuri PG (2020) SFLT-1 levels in COVID-19 patients: Association with outcome and thrombosis. Am J Hematol. https://doi.org/10.1002/ajh.26037
Mendoza M, Garcia-Ruiz I, Maiz N, Rodo C, Garcia-Manau P, Serrano B et al (2020) Pre-eclampsia-like syndrome induced by severe COVID-19: a prospective observational study. BJOG 127(11):1374–1380
Li L, Huang M, Shen J, Wang Y, Wang R, Yuan C et al (2021) Serum levels of soluble platelet endothelial cell adhesion molecule 1 in COVID-19 patients are associated with disease severity. J Infect Dis 223(1):178–179
Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P et al (2020) Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res 127(7):945–947
Borgel D, Chocron R, Grimaud M, Philippe A, Chareyre J, Brakta C et al (2021) Endothelial dysfunction as a component of SARS-CoV-2-related multisystem inflammatory syndrome in children with shock. Crit Care. https://doi.org/10.1097/CCM.0000000000005093
McCormack JJ, Lopes da Silva M, Ferraro F, Patella F, Cutler DF (2017) Weibel-palade bodies at a glance. J Cell Sci 130(21):3611-3617
Philippe A, Chocron R, Gendron N, Bory O, Beauvais A, Peron N et al (2021) Circulating Von Willebrand factor and high molecular weight multimers as markers of endothelial injury predict COVID-19 in-hospital mortality. Angiogenesis. https://doi.org/10.1007/s10456-020-09762-6
von Meijenfeldt FA, Havervall S, Adelmeijer J, Lundstrom A, Rudberg AS, Magnusson M et al (2021) Prothrombotic changes in patients with COVID-19 are associated with disease severity and mortality. Res Pract Thromb Haemost 5(1):132–141
Dupont A, Rauch A, Staessens S, Moussa M, Rosa M, Corseaux D et al (2021) Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler Thromb Vasc Biol 41(5):1760–1773
Bazzan M, Montaruli B, Sciascia S, Cosseddu D, Norbiato C, Roccatello D (2020) Low ADAMTS 13 plasma levels are predictors of mortality in COVID-19 patients. Intern Emerg Med 15(5):861–863
Blasi A, von Meijenfeldt FA, Adelmeijer J, Calvo A, Ibanez C, Perdomo J et al (2020) In vitro hypercoagulability and ongoing in vivo activation of coagulation and fibrinolysis in COVID-19 patients on anticoagulation. J Thromb Haemost 18(10):2646–2653
Rodriguez Rodriguez M, Castro Quismondo N, Zafra Torres D, Gil Alos D, Ayala R, Martinez-Lopez J (2021) Increased von Willebrand factor antigen and low ADAMTS13 activity are related to poor prognosis in covid-19 patients. Int J Lab Hematol. https://doi.org/10.1111/ijlh.13476
Escher R, Breakey N, Lammle B (2020) ADAMTS13 activity, von Willebrand factor, factor VIII and D-dimers in COVID-19 inpatients. Thromb Res 192:174–175
Turecek PL, Peck RC, Rangarajan S, Reilly-Stitt C, Laffan MA, Kazmi R et al (2021) Recombinant ADAMTS13 reduces abnormally up-regulated von Willebrand factor in plasma from patients with severe COVID-19. Thromb Res 201:100–112
Doevelaar AAN, Bachmann M, Holzer B, Seibert FS, Rohn BJ, Bauer F et al (2021) von Willebrand factor multimer formation contributes to immunothrombosis in coronavirus disease 2019. Crit Care Med 49(5):e512–e520
Mancini I, Baronciani L, Artoni A, Colpani P, Biganzoli M, Cozzi G et al (2021) The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost 19(2):513–521
Kwaan HC (2011) Microvascular thrombosis: a serious and deadly pathologic process in multiple diseases. Semin Thromb Hemost 37(8):961–978
Chavkin NW, Hirschi KK (2020) Single cell analysis in vascular biology. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2020.00042
Aird WC (2012) Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. 2(1):e006429
Liu J, Yuan L, Molema G, Regan E, Janes L, Beeler D et al (2011) Vascular bed-specific regulation of the von Willebrand factor promoter in the heart and skeletal muscle. Blood 117(1):342–351
Pusztaszeri MP, Seelentag W, Bosman FT (2006) Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem 54(4):385–395
Yamamoto K, de Waard V, Fearns C, Loskutoff DJ (1998) Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood 92(8):2791–2801
Cattaneo M, Bertinato EM, Birocchi S, Brizio C, Malavolta D, Manzoni M et al (2020) Pulmonary embolism or pulmonary thrombosis in COVID-19? Is the recommendation to use high-dose heparin for thromboprophylaxis justified? Thromb Haemost 120:1230–1232
Falasca L, Nardacci R, Colombo D, Lalle E, Di Caro A, Nicastri E et al (2020) Postmortem findings in italian patients with COVID-19: a descriptive full autopsy study of cases with and without comorbidities. J Infect Dis 222(11):1807–1815
Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P et al (2020) Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis 20(10):1135–1140
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383:120–128
Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C et al (2020) Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med 173(5):350–361
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Res 191:145-147
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MCA et al (2020) Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost 18(8):1995–2002
Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D (2020) COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med 201(10):1299–1300
Patel BV, Arachchillage D, A R, Bianchi P, Doyle JF, Garfield B, et al (2020) Pulmonary angiopathy in severe Covid-19: surrogate physiologic, imaging and hematologic observations. Am J Respirat Crit Care Med 202(5):690–699
Si-Mohamed S, Chebib N, Sigovan M, Zumbihl L, Turquier S, Boccalini S et al (2020) In vivo demonstration of pulmonary microvascular involvement in COVID-19 using dual-energy computed tomography. Eur Respir J 56(4):10
Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, Ferraz da Silva LF, Pierre de Oliveira E, Nascimento Saldiva PH et al (2020) Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thrombosis Haemostasis 18(6):1517–1519
The Severe Covid-19 GWAS Group (2020) Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med 383(16):1522–1534
O’Sullivan JM, Ward S, Fogarty H, O’Donnell JS (2020) More on “Association between ABO blood groups and risk of SARS-CoV-2 pneumonia.” Br J Haematol 190(1):27–28
Wu SC, Arthur CM, Wang J, Verkerke H, Josephson CD, Kalman D et al (2021) The SARS-CoV-2 receptor-binding domain preferentially recognizes blood group A. Blood Adv 5(5):1305–1309
Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I et al (2020) Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 58:102925
Belouzard S, Millet JK, Licitra BN, Whittaker GR (2012) Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4(6):1011–1033
Meduri GU, Annane D, Chrousos GP, Marik PE, Sinclair SE (2009) Activation and regulation of systemic inflammation in ARDS: rationale for prolonged glucocorticoid therapy. Chest 136(6):1631–1643
Yau JW, Teoh H, Verma S (2015) Endothelial cell control of thrombosis. BMC Cardiovasc Disord 15(1):130
Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ (2020) Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. 18(7):1548–1555
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. The Lancet 395(10234):1417–1418
Goldsmith CS, Miller SE, Martines RB, Bullock HA, Zaki SR (2020) Electron microscopy of SARS-CoV-2: a challenging task. The Lancet 395(10238):99
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M et al (2020) Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181(4):905–13.e7
Chen L, Li X, Chen M, Feng Y, Xiong C (2020) The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res 116(6):1097–1100
McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S et al (2021) Lack of evidence of ACE2 expression and replicative infection by SARSCoV-2 in human endothelial cells. Circulation 143(8):865–868
Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637
Karuppan MKM, Devadoss D, Nair M, Chand HS, Lakshmana MK (2021) SARS-CoV-2 infection in the central and peripheral nervous system-associated morbidities and their potential mechanism. Mol Neurobiol 13:1–16
Park EJ, Myint PK, Appiah MG, Darkwah S, Caidengbate S, Ito A, Matsuo E, Kawamoto E, Gaowa A, Shimaoka M (2021) The spike glycoprotein of SARS-CoV-2 binds to β1 integrins expressed on the surface of lung epithelial cells. Viruses 13(4):645. https://doi.org/10.3390/v13040645
Makowski L, Olson-Sidford W, Weisel JW (2021) Biological and clinical consequences of integrin binding via a rogue RGD motif in the SARS CoV-2 spike protein. Viruses 13(2):146. https://doi.org/10.3390/v13020146
Nolte MA, Nolte-'t ENM, Margadant HC (2021) Integrins control vesicular trafficking; new tricks for old dogs. Trends Biochem Sci 46(2):124–137. https://doi.org/10.1016/j.tibs.2020.09.001
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Investig 130(5):2620–2629
Reglero-Real N, Colom B, Bodkin JV, Nourshargh S (2016) Endothelial cell junctional adhesion molecules. Arterioscler Thromb Vasc Biol 36(10):2048–2057. https://doi.org/10.1161/ATVBAHA.116.307610
van der Bijl I, Nawaz K, Kazlauskaite U, van Stalborch A-M, Tol S, Jimenez A, van den Bout OI, Reinhard NR, Sonnenberg A, Margadant C (2020) Reciprocal integrin/integrin antagonism through kindlin-2 and Rho GTPases regulates cell cohesion and collective migration. Matrix Biol 93:60–78. https://doi.org/10.1016/j.matbio.2020.05.005
Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, Frye M, Zeuschner D, Nakayama M, Vestweber D, Adams RH (2015) Integrin β1 controls VE-cadherin localization and blood vessel stability. Nat Commun. https://doi.org/10.1038/ncomms7429
Rauch A, Dupont A, Goutay J, Caplan M, Staessens S, Moussa M et al (2020) Endotheliopathy is induced by plasma from critically Ill patients and associated with organ failure in severe COVID-19. Circulation 142(19):1881–1884
Polidoro RB, Hagan RS, de Santis SR, Schmidt NW (2020) Overview: systemic inflammatory response derived from lung injury caused by SARS-COV-2 infection explains severe outcomes in COVID-19. Front Immunol 11:1626
Teuwen L-A, Geldhof V, Pasut A, Carmeliet P (2020) COVID-19: the vasculature unleashed. Nat Rev Immunol 20(7):389–391
Arachchillage D, Shi C, Saliu D, Kozman P, Emma Mi4, Buti N, et al (2020) Efficacy and safety and of D-dimer, weight, and renal function adjusted thromboprophylaxis in patients with coronavirus disease 2019 (COVID-19). Semin Thromb Hemost 47(4):436-441
Paranjpe I, Fuster V, Lala A, Russak A, Glicksberg BS, Levin MA et al (2020) Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J Am College Cardiol. https://doi.org/10.1016/j.jacc.2020.05.001
Smadja DM, Gaussem P, Mauge L, Israel-Biet D, Dignat-George F, Peyrard S et al (2009) Circulating endothelial cells: a new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation 119(3):374–381
Chocron R, Galand V, Cellier J, Gendron N, Pommier T, Bory O et al (2021) Anticoagulation prior to hospitalization is a potential protective factor for COVID-19: insight from a French multicenter cohort study. J Am Heart Assoc. 10:1018288
Medcalf RL, Keragala CB, Myles PS (2020) Fibrinolysis and COVID-19: a plasmin paradox. J Thromb Haemost 18(9):2118–2122
Arachchillage DJ, Stacey A, Akor F, Scotz M, Laffan M (2020) Thrombolysis restores perfusion in COVID-19 hypoxia. Br J Haematol 190(5):e270–e274
Wu Y, Wang T, Guo C, Zhang D, Ge X, Huang Z et al (2020) Plasminogen improves lung lesions and hypoxemia in patients with COVID-19. QJM 113(8):539–545
Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M et al (2021) Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Science Reports 11(1):1580
Denorme F, Langhauser F, Desender L, Vandenbulcke A, Rottensteiner H, Plaimauer B et al (2016) ADAMTS13-mediated thrombolysis of t-PA–resistant occlusions in ischemic stroke in mice. Blood 127(19):2337–2345
Turecek P, Peck R, Rangarajan S, Reilly-Stitt C, Laffan M, Kazmi R et al (2021) Recombinant ADAMTS13 reduces abnormally up-regulated von Willebrand factor in plasma from patients with severe COVID-19. Thrombosis Research. 201:100–112
Meizlish ML, Goshua G, Liu Y, Fine R, Amin K, Chang E et al (2021) Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: a propensity score-matched analysis. Am J Hematol 96(4):471–479
Dutt T, Shaw RJ, Stubbs M, Yong J, Bailiff B, Cranfield T et al (2021) Real-world experience with caplacizumab in the management of acute TTP. Blood 137(13):1731–1740
Zheng L, Mao Y, Abdelgawwad MS, Kocher NK, Li M, Dai X et al (2016) Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Adv 1:75–83
Rahimi A, Samimagham HR, Azad MH, Hooshyar D, Arabi M, KazemiJahromi M (2021) The efficacy of N-Acetylcysteine in severe COVID-19 patients: A structured summary of a study protocol for a randomised controlled trial. Trials 22(1):271
Amado-Azevedo J, van Stalborch AD, Valent ET, Nawaz K, van Bezu J, Eringa EC et al (2021) Depletion of Arg/Abl2 improves endothelial cell adhesion and prevents vascular leak during inflammation. Angiogenesis. https://doi.org/10.1007/s10456-021-09781-x
Aman J, Duijvelaar E, Botros L, Kianzad A, J.R.Schippers, Smeele PJ, et al (2021) A randomised, double-blind, placebo controlled, clinical trial evaluating imatinib in patients with 5 severe COVID-19. Lancet Resp Med In Press
Noh JY, Yoon JG, Seong H, Choi WS, Sohn JW, Cheong HJ et al (2020) Asymptomatic infection and atypical manifestations of COVID-19: Comparison of viral shedding duration. J Infect 81(5):816–846
He X, Lau EHY, Wu P, Deng X, Wang J, Hao X et al (2020) Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat Med 26(5):672–675
Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z et al (2020) SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med 382(12):1177–1179
Zhao W, Zhong Z, Xie X, Yu Q, Liu J (2020) Relation between chest CT findings and clinical conditions of coronavirus disease (COVID-19) pneumonia: a multicenter study. AJR Am J Roentgenol 214(5):1072–1077
Salehi S, Abedi A, Balakrishnan S, Gholamrezanezhad A (2020) Coronavirus disease 2019 (COVID-19): a systematic review of imaging findings in 919 patients. AJR Am J Roentgenol 215(1):87–93
Ackermann M, Mentzer SJ, Jonigk D (2020) Pulmonary vascular pathology in Covid-19. Reply N Engl J Med 383(9):888–889
Ackermann M, Mentzer SJ, Jonigk D (2020) Visualization of SARS-CoV-2 in the lung. Reply N Engl J Med 383(27):2689–2690
Tobin MJ, Laghi F, Jubran A (2020) Why COVID-19 silent hypoxemia is baffling to physicians. Am J Respir Crit Care Med 202(3):356–360
Couzin-Frankel J (2020) The mystery of the pandemic’s “happy hypoxia.” Science 368(6490):455–456
Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN (2020) The pathophysiology of “happy” hypoxemia in COVID-19. Respir Res 21(1):198
Sylvester JT, Shimoda LA, Aaronson PI, Ward JP (2012) Hypoxic pulmonary vasoconstriction. Physiol Rev 92(1):367–520
Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J et al (2012) Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest 122(11):4218–4230
Herrmann J, Mori V, Bates JHT, Suki B (2020) Modeling lung perfusion abnormalities to explain early COVID-19 hypoxemia. Nat Commun 11(1):4883
Slessarev M, Cheng J, Ondrejicka M, Arntfield R (2020) Critical care western research G. Patient self-proning with high-flow nasal cannula improves oxygenation in COVID-19 pneumonia. Can J Anaesth 67(9):1288–1290
Caputo ND, Strayer RJ, Levitan R (2020) Early self-proning in awake, non-intubated patients in the emergency department: a single ED’s experience during the COVID-19 pandemic. Acad Emerg Med 27(5):375–378
Yang W, Cao Q, Qin L, Wang X, Cheng Z, Pan A et al (2020) Clinical characteristics and imaging manifestations of the 2019 novel coronavirus disease (COVID-19): a multi-center study in Wenzhou city, Zhejiang, China. J Infect 80(4):388–393
Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ et al (2020) Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther 5:33
Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D et al (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17(5):533–535
Caduff JH, Fischer LC, Burri PH (1986) Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat Rec 216(2):154–164
Filipovic N, Tsuda A, Lee GS, Miele LF, Lin M, Konerding MA et al (2009) Computational flow dynamics in a geometric model of intussusceptive angiogenesis. Microvasc Res 78(3):286–293
Lee GS, Filipovic N, Lin M, Gibney BC, Simpson DC, Konerding MA et al (2011) Intravascular pillars and pruning in the extraembryonic vessels of chick embryos. Dev Dyn 240(6):1335–1343
Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E et al (2012) Acute respiratory distress syndrome: the Berlin definition. JAMA 307(23):2526–2533
Ferguson ND, Fan E, Camporota L, Antonelli M, Anzueto A, Beale R et al (2012) The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med 38(10):1573–1582
Greene R, Lind S, Jantsch H, Wilson R, Lynch K, Jones R et al (1987) Pulmonary vascular obstruction in severe ARDS: angiographic alterations after i.v. fibrinolytic therapy. AJR Am J Roentgenol 148(3):501–508
Tomashefski JF Jr, Davies P, Boggis C, Greene R, Zapol WM, Reid LM (1983) The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol 112(1):112–126
Nuckton TJ, Alonso JA, Kallet RH, Daniel BM, Pittet JF, Eisner MD et al (2002) Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med 346(17):1281–1286
Chiumello D, Busana M, Coppola S, Romitti F, Formenti P, Bonifazi M et al (2020) Physiological and quantitative CT-scan characterization of COVID-19 and typical ARDS: a matched cohort study. Intensive Care Med 46(12):2187–2196
Goligher EC, Ranieri VM, Slutsky AS (2021) Is severe COVID-19 pneumonia a typical or atypical form of ARDS? And does it matter? Intensive Care Med 47(1):83–85
Beloncle FM, Pavlovsky B, Desprez C, Fage N, Olivier PY, Asfar P et al (2020) Recruitability and effect of PEEP in SARS-Cov-2-associated acute respiratory distress syndrome. Ann Intensive Care 10(1):55
Sinha P, Fauvel NJ, Singh P, Soni N (2013) Analysis of ventilatory ratio as a novel method to monitor ventilatory adequacy at the bedside. Crit Care 17(1):R34
Caviedes I, Soto R, Torres A (2021) Pulmonary angiopathy in severe COVID-19: physiological conclusions derived from ventilatory ratio? Am J Respir Crit Care Med 203(2):258–259
Carsetti A, Damiani E, Casarotta E, Scorcella C, Domizi R, Montomoli J, et al (2020) Sublingual microcirculation in patients with SARS-CoV-2 undergoing veno-venous extracorporeal membrane oxygenation. Microvasc Res 132:104064
Kanoore Edul VS, Caminos Eguillor JF, Ferrara G, Estenssoro E, Siles DSP, Cesio CE et al (2021) Microcirculation alterations in severe COVID-19 pneumonia. J Crit Care 61:73–75
do Espirito Santo DA, Lemos ACB, Miranda CH (2020) In vivo demonstration of microvascular thrombosis in severe COVID-19. J Thromb Thrombolysis 50(4):790–794
Ospina-Tascon GA, Bautista DF, Madrinan HJ, Valencia JD, Bermudez WF, Quinones E et al (2020) Microcirculatory dysfunction and dead-space ventilation in early ARDS: a hypothesis-generating observational study. Ann Intensive Care 10(1):35
Diehl JL, Peron N, Philippe A, Smadja DM (2020) Response to Damiani and colleagues. Ann Intensive Care 10(1):140
Patel BV, Arachchillage DJ, Ridge CA, Bianchi P, Doyle JF, Garfield B et al (2020) Pulmonary angiopathy in severe COVID-19: physiologic, imaging, and hematologic observations. Am J Respir Crit Care Med 202(5):690–699
Lang M, Som A, Mendoza DP, Flores EJ, Reid N, Carey D et al (2020) Hypoxaemia related to COVID-19: vascular and perfusion abnormalities on dual-energy CT. Lancet Infect Dis 20(12):1365–1366
Beenen LFM, Bos LD, Scheerder MJ, Lobe NHJ, Muller MCA, Schultz MJ et al (2020) Extensive pulmonary perfusion defects compatible with microthrombosis and thromboembolic disease in severe Covid-19 pneumonia. Thromb Res 196:135–137
Mauri T, Spinelli E, Scotti E, Colussi G, Basile MC, Crotti S et al (2020) Potential for lung recruitment and ventilation-perfusion mismatch in patients with the acute respiratory distress syndrome from coronavirus disease 2019. Crit Care Med 48(8):1129–1134
Sella N, Zarantonello F, Andreatta G, Gagliardi V, Boscolo A, Navalesi P (2020) Positive end-expiratory pressure titration in COVID-19 acute respiratory failure: electrical impedance tomography vs. PEEP/FiO2 tables. Crit Care. 24(1):540
Safaee Fakhr B, Araujo Morais CC, De Santis Santiago RR, Di Fenza R, Gibson LE, Restrepo PA et al (2020) Bedside monitoring of lung perfusion by electrical impedance tomography in the time of COVID-19. Br J Anaesth 125(5):e434–e436
Lerum TV, Aalokken TM, Bronstad E, Aarli B, Ikdahl E, Lund KMA et al (2021) Dyspnoea, lung function and CT findings 3 months after hospital admission for COVID-19. Eur Respir J 57(4):2003448
Mo X, Jian W, Su Z, Chen M, Peng H, Peng P et al (2020) Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur Respir J. 55(6):2001217
Dhawan RT, Gopalan D, Howard L, Vicente A, Park M, Manalan K et al (2021) Beyond the clot: perfusion imaging of the pulmonary vasculature after COVID-19. Lancet Respir Med 9(1):107–116
Chapman DG, Badal T, King GG, Thamrin C (2021) Caution in interpretation of abnormal carbon monoxide diffusion capacity in COVID-19 patients. Eur Respir J 57(1):2003263
Pfaender S, Mar KB, Michailidis E, Kratzel A, Boys IN, V’Kovski P et al (2020) LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat Microbiol 5(11):1330–1339
Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al (2020) Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 182(6):1401–18e18
Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, et al (2020) Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182(6):1419–40e23
Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM et al (2015) Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518(7540):542–546
Trumpp A, Essers M, Wilson A (2010) Awakening dormant haematopoietic stem cells. Nat Rev Immunol 10(3):201–209
Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ et al (2014) Clonal dynamics of native haematopoiesis. Nature 514(7522):322–327
Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q et al (2014) Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15(4):507–522
Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G et al (2016) Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 351(6269):aab2116
Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL (2019) The myeloid cell compartment-cell by cell. Annu Rev Immunol 37:269–293
Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD et al (2015) Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17(1):35–46
Herault A, Binnewies M, Leong S, Calero-Nieto FJ, Zhang SY, Kang YA et al (2017) Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544(7648):53–58
Boettcher S, Manz MG (2017) Regulation of inflammation- and infection-Driven Hematopoiesis. Trends Immunol 38(5):345–357
Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA (2010) Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465(7299):793–797
Manz MG, Boettcher S (2014) Emergency granulopoiesis. Nat Rev Immunol 14(5):302–314
Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA et al (2009) IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458(7240):904–908
Paschall AV, Zhang R, Qi CF, Bardhan K, Peng L, Lu G et al (2015) IFN regulatory factor 8 represses GM-CSF expression in T cells to affect myeloid cell lineage differentiation. J Immunol 194(5):2369–2379
Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S et al (2006) Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24(6):801–812
Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR et al (2014) Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 14(4):445–459
Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al (2017) Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21(2):225–40 e5
Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M et al (2012) Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol 188(12):5824–5828
Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al (2018) Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172(1–2):147–61e12
Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M et al (2014) Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 211(2):245–262
Kawano Y, Takaue Y, Hirao A, Abe T, Saito S, Matsunaga K et al (1991) Synergistic effect of recombinant interferon-gamma and interleukin-3 on the growth of immature human hematopoietic progenitors. Blood 77(10):2118–2121
Schurch CM, Riether C, Ochsenbein AF (2014) Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 14(4):460–472
Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A et al (2015) Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 347(6227):1260–1265
Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S et al (2016) Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol 18(6):607–618
Kandalla PK, Sarrazin S, Molawi K, Berruyer C, Redelberger D, Favel A et al (2016) M-CSF improves protection against bacterial and fungal infections after hematopoietic stem/progenitor cell transplantation. J Exp Med 213(11):2269–2279
Karmaus PWF, Herrada AA, Guy C, Neale G, Dhungana Y, Long L et al (2017) Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. J Exp Med 214(9):2629–2647
Mitroulis I, Chen LS, Singh RP, Kourtzelis I, Economopoulou M, Kajikawa T et al (2017) Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J Clin Invest 127(10):3624–3639
Vardhana SA, Wolchok JD (2020) The many faces of the anti-COVID immune response. J Exp Med 217(6):e20200678
Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al (2020) Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27(6):992–1000e3
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369(6504):718–724
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181(5):1036–45e9
Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M et al (2020) Immunology of COVID-19: current state of the science. Immunity 52(6):910–941
Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M et al (2020) Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584(7821):463–469
Liepelt A, Hohlstein P, Gussen H, Xue J, Aschenbrenner AC, Ulas T et al (2020) Differential gene expression in circulating CD14(+) monocytes indicates the prognosis of critically Ill patients with sepsis. J Clin Med 9(1):127
Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, Hung DT et al (2020) An immune-cell signature of bacterial sepsis. Nat Med 26(3):333–340
Mann ER, Menon M, Knight SB, Konkel JE, Jagger C, Shaw TN, et al (2020) Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci Immunol 5(51) :eabd6197
Meghraoui-Kheddar A, Barthelemy S, Boissonnas A, Combadiere C (2020) Revising CX3CR1 expression on murine classical and non-classical monocytes. Front Immunol 11:1117
Monneret G, Venet F (2016) Sepsis-induced immune alterations monitoring by flow cytometry as a promising tool for individualized therapy. Cytometry B Clin Cytom 90(4):376–386
Mengos AE, Gastineau DA, Gustafson MP (2019) The CD14(+)HLA-DR(lo/neg) monocyte: an immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front Immunol 10:1147
Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y et al (2014) Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care 18(4):R163
Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M et al (2010) Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med 207(7):1453–1464
Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al (2020) Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 181(7):1475–88 e12
Reyes M, Filbin MR, Bhattacharyya RP, Sonny A, Mehta A, Billman K, et al (2020) Induction of a regulatory myeloid program in bacterial sepsis and severe COVID-19. bioRxiv
Psaila B, Mead AJ (2019) Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 133(13):1427–1435
Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL et al (2013) Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154(5):1112–1126
Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H et al (2015) Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell 17(4):422–434
Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM et al (2017) The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544(7648):105–109
Roncati L, Ligabue G, Nasillo V, Lusenti B, Gennari W, Fabbiani L et al (2020) A proof of evidence supporting abnormal immunothrombosis in severe COVID-19: naked megakaryocyte nuclei increase in the bone marrow and lungs of critically ill patients. Platelets 31(8):1085–1089
Valdivia-Mazeyra MF, Salas C, Nieves-Alonso JM, Martin-Fragueiro L, Barcena C, Munoz-Hernandez P et al (2021) Increased number of pulmonary megakaryocytes in COVID-19 patients with diffuse alveolar damage: an autopsy study with clinical correlation and review of the literature. Virchows Arch 478(3):487–496
Bernardes JP, Mishra N, Tran F, Bahmer T, Best L, Blase JI, et al (2020) Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity 53(6):1296–314e9
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E et al (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377(2):111–121
Duployez N, Demonchy J, Berthon C, Goutay J, Caplan M, Moreau AS et al (2020) Clinico-biological features and clonal hematopoiesis in patients with severe COVID-19. Cancers (Basel). 12(7):1992
Bolton KL, Koh Y, Foote MB, Im H, Jee J, Sun CH, et al (2020) Clonal hematopoiesis is associated with risk of severe Covid-19. medRxiv
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X et al (2015) Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525(7569):389–393
Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al (2018) Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell 23(6):833–49e5
Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y et al (2018) Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554(7690):123–127
Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW et al (2014) Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19(5):821–835
Garcia Luis, de Guadiana R, Mulero MDR, Olivo MH, Rojas CR, Arenas VR, Morales MG et al (2021) Circulating levels of GDF-15 and calprotectin for prediction of in-hospital mortality in COVID-19 patients: A case series. J Infect 82(2):e40–e2
Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y et al (2020) Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol Immunol 17(9):992–994
Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA et al (2021) Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol 109(1):67–72
Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J (2018) S100A8/A9 in Inflammation. Front Immunol 9:1298
Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T et al (2017) S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol 18(6):622–632
Oduro KA Jr, Liu F, Tan Q, Kim CK, Lubman O, Fremont D et al (2012) Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood 120(11):2203–2213
Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK et al (2020) Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation 141(13):1080–1094
Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E et al (2017) Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest 127(6):2133–2147
Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C et al (2016) Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med 22(3):288–297
Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L et al (2016) The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 128(25):2960–2975
Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N et al (2013) Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest 123(11):4595–4611
Cheng P, Eksioglu EA, Chen X, Kandell W, Le Trinh T, Cen L et al (2019) S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia 33(8):2034–2046
Leimkuhler NB, Gleitz HFE, Ronghui L, Snoeren IAM, Fuchs SNR, Nagai JS, et al (2021) Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell 28(4):637–52 e8.
Ratajczak MZ, Ratajczak J, Suszynska M, Miller DM, Kucia M, Shin DM (2017) A novel view of the adult stem cell compartment from the perspective of a quiescent population of very small embryonic-like stem cells. Circ Res 120(1):166–178
Mancuso P, Gidaro A, Gregato G, Raveane A, Cremonesi P, Quarna J et al (2020) Circulating endothelial progenitors are increased in COVID-19 patients and correlate with SARS-CoV-2 RNA in severe cases. J Thromb Haemost 18(10):2744–2750
Guerin CL, Guyonnet L, Goudot G, Revets D, Konstantinou M, Chipont A et al (2020) Multidimensional proteomic approach of endothelial progenitors demonstrate expression of KDR restricted to CD19 cells. Stem Cell Rev Rep. https://doi.org/10.1007/s12015-020-10062-1
Qu J, Wu C, Li X, Zhang G, Jiang Z, Li X et al (2020) Profile of immunoglobulin G and IgM antibodies against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis 71(16):2255–2258
Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK et al (2020) Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med 26(6):845–848
Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y et al (2020) Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin Infect Dis 71(16):2027–2034
Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J et al (2020) Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med 26(8):1200–1204
McAndrews KM, Dowlatshahi DP, Dai J, Becker LM, Hensel J, Snowden LM et al (2020) Heterogeneous antibodies against SARS-CoV-2 spike receptor binding domain and nucleocapsid with implications for COVID-19 immunity. JCI Insight. https://doi.org/10.1172/jci.insight.142386
Phipps WS, SoRelle JA, Li QZ, Mahimainathan L, Araj E, Markantonis J et al (2020) SARS-CoV-2 antibody responses do not predict COVID-19 disease severity. Am J Clin Pathol 154(4):459–465
Lynch KL, Whitman JD, Lacanienta NP, Beckerdite EW, Kastner SA, Shy BR et al (2021) Magnitude and kinetics of anti-severe acute respiratory syndrome coronavirus 2 antibody responses and their relationship to disease severity. Clin Infect Dis 72(2):301–308
Hu WT, Howell JC, Ozturk T, Benameur K, Bassit LC, Ramonell R et al (2020) Antibody profiles according to mild or severe SARS-CoV-2 Infection, Atlanta, Georgia, USA, 2020. Emerg Infect Dis 26(12):2974–2978
Young BE, Ong SWX, Ng LFP, Anderson DE, Chia WN, Chia PY et al (2020) Viral dynamics and immune correlates of COVID-19 disease severity. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa1280
Amrun SN, Lee CY, Lee B, Fong SW, Young BE, Chee RS et al (2020) Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine 58:102911
Ren L, Fan G, Wu W, Guo L, Wang Y, Li X et al (2021) Antibody responses and clinical outcomes in adults hospitalized with severe coronavirus disease 2019 (COVID-19): a post hoc analysis of LOTUS China trial. Clin Infect Dis 72(10):e545–e551
Wang Y, Zhang L, Sang L, Ye F, Ruan S, Zhong B et al (2020) Kinetics of viral load and antibody response in relation to COVID-19 severity. J Clin Invest 130(10):5235–5244
Casadevall A, Joyner MJ, Pirofski LA (2020) SARS-CoV-2 viral load and antibody responses: the case for convalescent plasma therapy. J Clin Invest 130(10):5112–5114
Luo YR, Chakraborty I, Yun C, Wu AHB, Lynch KL (2020) Kinetics of SARS-CoV-2 antibody avidity maturation and association with disease severity. Clin Infect Dis 10:2. https://doi.org/10.1093/cid/ciaa1389
Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J et al (2020) Treatment of 5 critically Ill patients with COVID-19 with convalescent plasma. JAMA 323(16):1582–1589
Szwebel TA, Veyer D, Robillard N, Eshagh D, Canoui E, Bruneau T et al (2021) Usefulness of plasma SARS-CoV-2 RNA quantification by droplet-based digital PCR to monitor treatment against COVID-19 in a B-cell lymphoma patient. Stem Cell Rev Rep 17(1):296–299
Hueso T, Pouderoux C, Pere H, Beaumont AL, Raillon LA, Ader F et al (2020) Convalescent plasma therapy for B-cell-depleted patients with protracted COVID-19. Blood 136(20):2290–2295
Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P et al (2020) Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ 371:m3939
Simonovich VA, Burgos Pratx LD, Scibona P, Beruto MV, Vallone MG, Vazquez C et al (2021) A randomized trial of convalescent plasma in Covid-19 severe pneumonia. N Engl J Med 384(7):619–629
Huijbers EJM, van Beijnum JR, Le CT, Langman S, Nowak-Sliwinska P, Mayo KH et al (2018) An improved conjugate vaccine technology; induction of antibody responses to the tumor vasculature. Vaccine 36(21):3054–3060
Huijbers EJM, Griffioen AW (2017) The revival of cancer vaccines—the eminent need to activate humoral immunity. Hum Vaccin Immunother 13(5):1112–1114
Smadja DM, Yue QY, Chocron R, Sanchez O, Lillo-Le Louet A (2021) Vaccination against COVID-19: insight from arterial and venous thrombosis occurrence using data from VigiBase. Eur Respir J. https://doi.org/10.1183/13993003.00956-2021
Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S (2021) Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med 384(22):2092–2101
Schultz NH, Sorvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT et al (2021) Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med 384(22):2124–2130
Funding
We thank Maud BARRANDON for Fig. 1 design. DM SMADJA, O SANCHEZ and JL DIEHL are funded with grants from French national agency for research ANR Flash-COVID SARCODO (Fondation de France), ANR AAP RA-COVID-19 VASCOV (CAPNET label "priorité nationale de recherche" by French Government), Mécénat Covid AP-HP and SARCODO-EP (COV21033- Contrat de recherche clinique DRCI AP-HP). M FONTENAY work is funded by the French national agency for research ANR AAP RA-COVID-19 CALPRO, the Mécénat Covid AP-HP and the Site de Recherche Intégré sur le Cancer CARPEM of the Institut National du Cancer. AW GRIFFIOEN and EJM HUIJBERS are funded by the Dutch Cancer Society (KWF2018–11651). P NOWAK-SLIWINSKA is funded by the European Research Council (ERC-680209) and by the Swiss National Science Foundation (#310030–197878). AM RANDI is funded by the British Heart Foundation (BHF) and the Imperial College COVID response Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
CK reports personal fees from Maquet, Xenios, and Bayer, as well as non-financial support from Speaker of the German register of ICUs and grants from the German Ministry of Research and Education.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Smadja, D.M., Mentzer, S.J., Fontenay, M. et al. COVID-19 is a systemic vascular hemopathy: insight for mechanistic and clinical aspects. Angiogenesis 24, 755–788 (2021). https://doi.org/10.1007/s10456-021-09805-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-021-09805-6