
Abstract
Following the SARS outbreak, extensive surveillance was undertaken globally to detect and identify coronavirus diversity in bats. This study sought to identify the diversity and prevalence of coronaviruses in bats in the Australasian region. We identified four different genotypes of coronavirus, three of which (an alphacoronavirus and two betacoronaviruses) are potentially new species, having less than 90% nucleotide sequence identity with the most closely related described viruses. We did not detect any SARS-like betacoronaviruses, despite targeting rhinolophid bats, the putative natural host taxa. Our findings support the virus-host co-evolution hypothesis, with the detection of Miniopterus bat coronavirus HKU8 (previously reported in Miniopterus species in China, Hong Kong and Bulgaria) in Australian Miniopterus species. Similarly, we detected a novel betacoronavirus genotype from Pteropus alecto which is most closely related to Bat coronavirus HKU9 identified in other pteropodid bats in China, Kenya and the Philippines. We also detected possible cross-species transmission of bat coronaviruses, and the apparent enteric tropism of these viruses. Thus, our findings are consistent with a scenario wherein the current diversity and host specificity of coronaviruses reflects co-evolution with the occasional host shift.
Similar content being viewed by others


Introduction
Novel coronaviruses were responsible for the global outbreak of severe acute respiratory syndrome (SARS) in 2003–2004, and for the ongoing sporadic outbreaks of Middle East respiratory syndrome (MERS), first described in 2012 (Drosten et al. 2003; Zaki et al. 2012). Bats have been identified as the natural hosts of the progenitor viruses of both SARS and MERS coronaviruses (Lau et al. 2005b; Li et al. 2005; Ge et al. 2013; Memish et al. 2013). Following the SARS outbreak, extensive surveillance was undertaken globally to detect and identify coronavirus diversity in bats, given their evident potential to host novel zoonotic coronaviruses (Woo et al. 2006). The majority of these studies were based on reverse-transcriptase PCR detection of coronavirus RNA in bat faeces and tissues, or serological detection of coronavirus antibodies in blood samples. Coronaviruses have been detected in 11 of the 18 extant families of bats in more than 25 countries in Asia, Africa, Europe and the Americas (Drexler et al. 2014).
Coronaviruses are enveloped positive-sense RNA viruses. Their taxonomy has undergone major revision over the past decade, primarily because of the identification of a suite of novel coronaviruses in bats. The family Coronaviridae is now divided into the two subfamilies of Coronavirinae and Torovirinae, with the former containing the genera Alpha-, Beta-, Gamma- and Deltacoronavirus (International Committee on Taxonomy of Viruses 2011). SARS and MERS coronaviruses belong to the genus Betacoronavirus, with all coronaviruses detected in bats being either alpha- or betacoronaviruses (Drexler et al. 2014).
Bat coronaviruses have a narrow host range and are typically species, genus or family-specific, independent of location (Poon et al. 2004; Chu et al. 2006; Tang et al. 2006; Woo et al. 2006; Gloza-Rausch et al. 2008; Pfefferle et al. 2009; Drexler et al. 2014). Drexler et al. (2010) hypothesised that this virus-host specificity could be used in a prospective manner to predict the geographic distribution of various bat coronavirus species, and showed that the same alphacoronavirus (Miniopterus bat coronavirus HKU8), which had previously been reported in Miniopterus species in China and Hong Kong, occurred in M. schreibersii in Bulgaria, over 8000 km distant. Further, related coronaviruses hosted by bats of the same family have been detected in Vespertilionidae (Cui et al. 2007) and Rhinolophidae bats (Lau et al. 2005b; Li et al. 2005).
The diversity and prevalence of coronaviruses in bats in the Australasian region has not yet been reported. Given the zoonotic potential of bat coronaviruses, a better understanding of the distribution and ecology of these viruses is essential to identify any potential threat to human health and to inform biosecurity preparedness. With this aim, we undertook a virological and serological survey of various bat populations in Australia and neighbouring countries for evidence of coronavirus infection. We focused primarily on insectivorous bats, which reportedly host the largest diversity of bat coronaviruses, and particularly targeted bats of the genus Rhinolophus, recognised as hosting SARS-like coronaviruses. We also compared relative detection frequency in various potential routes of excretion.
Materials and Methods
Study Samples
Faecal samples, anal swabs or intestinal content samples were obtained by either primary fieldwork or sourced from archived samples.
Fieldwork: Bat Capture and Sample Collection
Fieldwork was undertaken at multiple locations in Australia and Taiwan between 2006 and 2009, using harp traps to capture insectivorous bats. Particular effort was made to capture bats at locations where rhinolophid bats were previously known to be present. Captured bats were individually housed in clean cloth bags and a polystyrene cooler prior to sample collection. Bat identification was based on morphological characteristics (Churchill 2008) and was undertaken by experienced biologists. Occasionally, bats were only identified to genus level. All bats were released at their point of capture within 6 h. A single faecal pellet (collected directly from a defecating bat or from its clean cloth bag) was placed into 1 ml of sucrose potassium glutamate albumin (SPGA) with added penicillin, streptomycin and fungizone. When no faecal pellet was obtained, the anus was swabbed and the swab placed in 1 ml SPGA, and stored at 4°C prior to RNA extraction. Faecal samples collected from bats in Taiwan were placed into 1 ml of AVL buffer from the QIAamp® Viral RNA Mini Kit (QIAGEN) and stored at room temperature for 1 week prior to extraction. Faecal swabs from 33 Pteropus alecto and 27 P. poliocephalus opportunistically obtained from colleagues conducting concurrent henipavirus research in south-east Queensland in 2009 were placed in 1 ml SPGA and stored at 4°C prior to extraction.
Bats were bled as described by Smith et al. (2010), and blood was diluted 1:10 in phosphate-buffered saline. In addition to faecal and blood samples, oropharyngeal swabs were collected from 30 bats (14 Miniopterus australis, 16 M. schreibersii) from south-east Queensland.
Bat capture and sample collection was undertaken in direct accordance with animal ethics committee permits SA 2006/06/117 and SA 2007/005/194 issued by the (then) Queensland Department of Primary Industries and Fisheries, and scientific purposes permits WISP03887606 and WISP04906107 issued by the Queensland Parks and Wildlife Service of the (then) Queensland Environmental Protection Agency.
Archived Samples
Twenty archival samples of intestines (stored at −70°C) from bats caught in central Queensland in 1996 were homogenised (2 mm2 section) in 1 ml of SPGA. In addition, archived (−20°C) serum samples collected from various bat species between 1999 and 2006 by colleagues in the course of henipavirus and other surveillance were serologically screened (Western Australia, n = 119; Northern Territory, n = 40; East Timor, n = 36; Indonesia, n = 67; Malaysia, n = 101 and Papua New Guinea, n = 65). Archived samples were sourced from the Queensland Department of Agriculture and Fisheries Biological Sciences Laboratory (BSL) in Brisbane, Australia.
Coronavirus RNA Detection and Sequencing
Faecal pellets, intestinal samples, anal and oropharyngeal swabs in SPGA were centrifuged as required, and RNA was extracted from 560 μl of the supernatant using the QIAamp® Viral RNA Mini Kit (QIAGEN) following the manufacturer’s instructions. RNA was similarly extracted from 140 μl of diluted blood. Reverse transcription PCR (RT-PCR) targeting a conserved region of the coronavirus RNA-dependent RNA polymerase (RdRp) gene, as described by Poon et al. (2005), was performed using the Superscript III One-Step RT-PCR System with Platinum® Taq DNA Polymerase (Invitrogen). Amplicons consistent with the expected length of 440 bp were purified using the QIAquick® PCR Purification Kit (QIAGEN) as per the manufacturer’s instructions. Purified amplicons were directly sequenced in both directions using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) as per the manufacturer’s instructions. The extension products were purified using the ethanol/EDTA precipitation method (Applied Biosystems 2002) and analysed at the Griffith University DNA Sequencing Facility (Brisbane, Australia). Nucleotide sequence traces were edited using Sequence Scanner v1.0 (Applied Biosystems).
Molecular Phylogenetic Analysis
Phylogenetic analysis of a 440 bp sequence from the RNA-dependent RNA polymerase (RdRp) gene from representative sequences from this study, and from 33 other sequences including the ICTV reference coronaviruses, was conducted using the Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar 2000). Initial tree(s) for the heuristic search were obtained by applying the Neighbour-Joining method to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach. A discrete Gamma distribution was used to model evolutionary rate differences amongst sites [5 categories (+G, parameter = 0.6965)]. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 0.0000% sites). Evolutionary analyses were conducted in MEGA6 (Tamura et al. 2013).
Anti-coronavirus Antibody Detection
The archived serum samples were screened for cross-reactive antibodies to SARS or SARS-like viruses using a SARS coronavirus crude antigen ELISA modified from the original protocol developed by Yu et al. (2008). Briefly, total viral antigen was used to coat a 96-well Nunc Max ELISA plate in 0.1 M carbonate coating buffer, followed by incubation for 30 min (all incubation was done at 37°C). After washing with PBST buffer, the plate was incubated for 30 min with blocking buffer (2% skimmed milk in PBST). Testing sera were diluted 1:50 in blocking buffer and incubated for 30 min, followed by washing with PBST. HRP-conjugated Protein-A/G (1:1 mix, ZYMED Laboratories) was used to detect the bound bat antibodies by incubation for 30 min with a 1:2000 diluted solution. After washing, TMB substrate was added for colour development and the reading was taken after 10 min at 450 nm using an ELISA plate reader (Multiskan Ascent, Thermo Labsystems).
Prevalence Estimation
Prevalence of coronavirus RNA and coronavirus antibodies was estimated for species and country of sample origin (in Australia, for species and state/territory of sample origin), with 95% confidence intervals calculated using the Jeffreys method (Brown et al. 2001).
Results
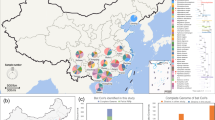
Field and archived samples were derived from 2195 bats from Australia, Papua New Guinea, East Timor, Indonesia, Malaysia and Taiwan (Fig. 1). The sampled bats belonged to 22 different genera within eight families (Table 1). In total, 1484 faecal/anal swab samples, 30 blood samples and 30 oropharyngeal swab samples from captured bats, and 20 archived bat intestinal samples were subjected to RT-PCR. Serology was performed on a total 1808 serum samples (Table 1).

Sampling locations. Locations of 2195 bats sampled between 1996 and 2009 and screened for evidence of coronavirus infection, comprising bats from south-east Queensland (SEQ, n = 1162), central Queensland (CQ, n = 42), far-north Queensland (FNQ, n = 222), the Northern Territory (NT, n = 333), Western Australia (WA, n = 119), Taiwan (n = 43), East Timor (n = 36), Indonesia (n = 67), Malaysia (n = 101) and Papua New Guinea (n = 65).
Detection of Coronavirus RNA and Antibodies
Coronavirus RNA was detected in faecal, intestinal or anal swab samples of seven species of Australian bats from five families (Table 1). RNA-positive bats were found in the three broad sampling locations in Queensland and the Northern Territory, but not in Western Australia. Viral RNA prevalence in the seven positive species ranged from 1/126 (0.8%, 95% CI 0.1–3.6%) in Rhinonicteris aurantia to 14/30 (46.7%, 95% CI 29.8–64.1%) in Miniopterus australis. Limited sample size (and consequent wide 95% confidence intervals) constrained statistical confidence in prevalence estimates for some species in some locations.
In the 30 bats from which faecal samples, blood and oropharyngeal swabs were collected, coronavirus RNA was detected in 11 faecal samples and two oropharyngeal samples, the latter from bats with RNA-positive faecal samples. No blood samples yielded viral RNA.
Coronavirus antibodies were detected in 23 species of bats from Australia, East Timor, Indonesia, Malaysia and Papua New Guinea (Table 1). In Australia, antibody-positive bats were found in all five broad sampling locations, including remote Western Australia. Further, anti-coronavirus antibodies were detected in five of six species in which coronavirus RNA was detected. The seventh RNA-positive species did not have serum available for testing. As with viral RNA prevalence, seroprevalence varied markedly between species and locations: the highest seroprevalence estimate was 100% on three occasions, namely Scotorepens spp. from south-east Queensland (24/24, 100% 95% CI 90.2–100%), Scotophilus spp. from Malaysia (4/4, 95% CI 55.5–100%) and Miniopterus australis from Western Australia (1/1, 95% CI 14.7–100%); the lowest detected seroprevalence was 1/63 (1.6%, 95% CI 0.2–7.2%) in Macroderma gigas in the Northern Territory, Australia.
Sequencing and Phylogenetic Analysis
The phylogenetic analyses identified four different coronavirus genotypes amongst the sequenced samples. Two genotypes clustered in the genus Alphacoronavirus and two clustered in the genus Betacoronavirus. Significantly, three of the four genotypes shared less than 90% nucleotide sequence identity with the most closely related known coronaviruses. The tree with the highest log likelihood is shown in Fig. 2. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The final consensus sequences, derived from sense and anti-sense primers, were deposited in GenBank under accessions numbers EU834950-EU834956.

Phylogenetic analysis of nucleotide sequences identified in Australian bats. Phylogenetic analysis of the 440 bp sequence from the RNA-dependent RNA polymerase (RdRp) gene from ten representative sequences from this study and 33 other sequences was conducted using the Maximum Likelihood method. The tree with the highest log likelihood (−16,168.6385) is shown. Values to the left of branches are the percentage of trees in which the associated taxa clustered. The tree is drawn to scale, with branch lengths indicating the number of substitutions per site. Coronavirus sequences identified in this study are shown in bold. Square brackets are used to identify coronavirus species and genus groups. Coronavirus nomenclature: Host species/country of origin/laboratory identification/year collected (GenBank accession).
The most widespread genotype, both in terms of host species and geographical location, was an alphacoronavirus that shared greater than 99% nucleotide sequence identity with the ICTV reference virus Miniopterus bat coronavirus HKU8. This genotype was identified in M. australis in central Queensland in 1996, and in south-east Queensland and far-north Queensland between 2006 and 2008; in M. schreibersii from south-east Queensland and the Northern Territory sampled between 2006 and 2008; and in a single Rhinolophus megaphyllus from far-north Queensland sampled in 2007 (Table 1).
A second and novel alphacoronavirus genotype was identified in Myotis macropus and Vespadelus pumilus from south-east Queensland (Table 1; Fig. 2). This nucleotide sequence shares only 89% identity with any reported coronavirus sequence, and is most closely related to a putative coronavirus species identified in Pipistrellus kuhlii from Italy and Spain (Lelli et al. 2013).
A novel betacoronavirus identified in a single Rhinonicteris aurantia from the Northern Territory was most closely related to one identified in Hipposideros caffer ruber from Ghana (Table 1; Fig. 2), although the two sequences had less than 81% identity. A second novel betacoronavirus genotype was identified in Pteropus alecto in south-east Queensland; the most closely related sequences are coronaviruses detected in Rousettus aegyptiacus from Kenya and Cynopterus brachyotis from the Philippines (Table 1; Fig. 2), although nucleotide identity was less than 87%.
Discussion
This is the first report of detection of coronavirus RNA and anti-coronavirus antibodies in bats in the Australasian region, and illustrates that bats in the region are host to a range of coronaviruses. The prevalence of each varied across species and location, with the prevalence of either viral RNA or antibodies being relatively high in some populations. This variability highlights the complexity of exposure, infection and immune dynamics over space and time. In two species with relatively large sample sizes (Miniopterus australis and M. schreibersii in south-east Queensland), 25% of 154 individuals and 27% of 238 individuals, respectively, were positive for coronavirus RNA. Studies elsewhere have shown similar prevalence rates (Tsuda et al. 2012; Suzuki et al. 2014); others have reported much lower prevalence (Anthony et al. 2013). Seroprevalence in our study was also high in some populations, including those that were negative for viral RNA. This antibody-positive, viral RNA-negative finding most plausibly reflects previous infection. Alternatively, virus excretion associated with current infection may have been occurring at a concentration below the detection level of the RT-PCR assay. In some cases, the limited sample size may have constrained detection of viral RNA in the sampled population. Although there have been fewer serological studies than molecular studies in bats, others have also found high coronavirus antibody prevalence in some bat species, albeit using a different serological assay (Tsuda et al. 2012). Indeed, given that our ELISA was based on SARS-CoV antigens, it may be that broader coronavirus seroprevalence is underestimated.
Our molecular analysis was based on highly conserved 440-bp sequence of the coronavirus RdRp gene, a common length and site for sequence data analyses conducted in many other bat coronavirus studies. Although constrained by the short length of sequence, we were able to identify four different genotypes of coronavirus, three of which (an alphacoronavirus and two betacoronaviruses) are potentially new species, having less than 90% nucleotide sequence identity with the most closely related described viruses. To obtain a surrogate estimation of taxonomy, Drexler et al. (2010) proposed a scheme based on 816-bp sequence of the RdRp gene to form RdRp grouping units (RGU), defining species separation in mammalian coronaviruses, with >4.8% amino acid sequence distance for alphacoronaviruses and >6.3% amino acid sequence distance for betacoronaviruses. While our field and laboratory work predated this publication and thus precluded use of the approach, based on nucleotide sequence identity, the three novel coronavirus genotypes identified in our study may represent new species. Regardless, our findings support the hypothesis of Woo et al. (2006) that bats host a large diversity of novel coronaviruses.
Despite targeting rhinolophid bats, the putative natural host taxa of SARS coronaviruses (Lau et al. 2005a; Li et al. 2005), and testing faecal samples from more than 500 individuals from multiple locations and detecting alphacoronavirus RNA, we did not detect any SARS-like betacoronaviruses. We did detect anti-coronavirus antibodies in a small percentage of R. megaphyllus, but we believe this more plausibly reflects alphacoronavirus exposure, and thus have no robust indication that SARS-like coronaviruses circulate in Australian bats.
Previous studies have reported that bat coronaviruses have a relatively narrow host range, independent of location (Poon et al. 2004; Chu et al. 2006; Tang et al. 2006; Woo et al. 2006; Gloza-Rausch et al. 2008; Pfefferle et al. 2009; Drexler et al. 2014). Our study supports this host species-virus association with the identification of Miniopterus bat coronavirus HKU8 (previously reported in Miniopterus species in China, Hong Kong and Bulgaria) in M. australis and M. schreibersii from Australia, some 7,000 km from Hong Kong and some 15,000 km from Bulgaria. Similarly, the novel betacoronavirus genotype from Rhinonicteris aurantia is most closely related to betacoronavirus sequence identified in Ghana from bats of the same family Hipposideridae (Pfefferle et al. 2009), although the lower sequence identity should be noted. Finally, our betacoronavirus genotype from P. alecto is most closely related to Bat coronavirus HKU9, identified in Rousettus spp from China and Kenya, and Cynopterus brachyotis from the Philippines, all of which belong to the family Pteropodidae. Notwithstanding the limited number of sequences and modest size of the amplicon evaluated, our findings support the contention that, regardless of location, bats belonging to the same species, genus or family are likely to host closely related coronaviruses. With the recent revision of the taxonomy of bats using comparative-method and molecular studies (Hutcheon and Kirsch 2006), the suborder Pteropodiformes now comprises, amongst others, bats from the families Hipposideridae, Rhinolophidae and Pteropodidae. To date, betacoronaviruses have been identified predominantly from bats of these families, so it appears that the host-virus association of bat coronaviruses may extend to bats of the same suborder. Thus, as yet unidentified betacoronaviruses may still be found in other species in other families (Craseonycteridae, Megadermatidae and Rhinopomatidae) in the suborder Pteropodiformes.
Our finding that an alphacoronavirus Miniopterus bat coronavirus HKU8 detected in a Rhinolophus megaphyllus bat was genetically identical to that identified in 50% of M. australis bats at the same roost site (Table 1) suggests that a cross-species transmission event has occurred. The detection of viral RNA in only one of the 58 R. megaphyllus sampled at that site may indicate that only sporadic spillover is occurring, and intra-species transmission in R. megaphyllus is not (yet) occurring. The genus Rhinolophus is hypothesised to be more likely to foster host shifts and foster the emergence of coronaviruses than other genera (Cui et al. 2007), and our observation is consistent with this. Similarly, our detection of coronavirus RNA in one of the 126 R. aurantia in the absence of any seropositivity in the population may also suggest transmission from another bat species. The sampled R. aurantia population was co-roosting with populations of Hipposideros ater and Macroderma gigas bats (Fig. 3), both of which were positive for anti-coronavirus antibodies, and although negative for coronavirus RNA at the time of sampling, constitute a plausible source of infection. Further investigation of these two species, beyond the scope of this study, is necessary to test this hypothesis. The alternative interpretation of the single RNA detection in both R. megaphyllus and R. aurantia is that the PCR results are spurious. However, we believe that the rigour of our field and laboratory techniques make cross-contamination of samples highly unlikely.
Two methods of evolution have been proposed to explain the genetic diversity of bat coronaviruses and their host specificity (Cui et al. 2007; Vijaykrishna et al. 2007). Divergent evolution requires the cross-species transmission of a common ancestor virus and subsequent adaption and establishment in the new host. To account for the identification of related coronaviruses in related species of bats in different locations throughout the world, divergent evolution requires the global distribution of each newly diverged coronavirus. This process is theoretically possible for those bat species that exhibit long-range nomadic movement (Breed et al. 2010). The second evolutionary method is the co-evolution of bat species and their coronaviruses (Cui et al. 2007), whereby the divergence of each bat species is mirrored by the divergence of the coronavirus it hosts. This method accounts for the diversity, relatedness and global distribution of bat coronaviruses, but would require the age of bat coronaviruses to parallel that of the most common bat ancestor, i.e. 65 million years (Churchill 2008). In addition, co-evolution alone does not explain the presence of different coronavirus genera in the same species or genus; for example, hipposiderid and rhinolophid bats hosting both alpha- and betacoronaviruses (Woo et al. 2006; Pfefferle et al. 2009), which intuitively appear to require episodes of host-shifting. Our findings are consistent with a scenario wherein the current diversity of coronaviruses and host specificity in Australasian bats reflects co-evolution with the occasional host shift, most plausibly by hipposiderid and/or rhinolophid bats (Cui et al. 2007). The significance of host geography and host ecology on virus evolution and host-virus interactions has recently been canvassed for bat rabies virus (Streicker et al. 2010, 2012). The concepts may be equally relevant for coronaviruses across the geographic and taxonomic spectrum, and warrant future investigation beyond the scope of this regional study.
The majority of coronaviruses previously reported in bats were detected in faecal samples or rectal swabs indicating a predominantly enteric tropism (Lau et al. 2005a; Poon et al. 2005; Chu et al. 2006; Tang et al. 2006; Dominguez et al. 2007; Lau et al. 2007). From the (albeit limited) number of bats from which additional sample types were tested in this study (n = 30), coronavirus RNA was detected in oropharyngeal swabs (n = 2) only from animals that were RNA-positive in faecal samples (n = 11), supporting a predominantly enteric tropism of bat coronaviruses. Corroborative studies are needed given our limited sample size. Lack of viral RNA detection in blood samples indicates that blood is not useful for the detection of bat coronaviruses.
Conclusion
This study identified coronavirus RNA and antibodies in a range of bat taxa in Australia and the neighbouring countries of East Timor, Indonesia, Malaysia and Papua New Guinea. Four different coronavirus genotypes were identified, including one alphacoronavirus and two betacoronaviruses that are evidently novel. We also detected possible cross-species transmission of bat coronaviruses, and provide further support for the apparent enteric tropism of these viruses. The findings advance our knowledge of the diversity of coronaviruses in bats. This diversity, the global distribution of bats, and the propensity of coronaviruses to successfully cross species barriers, suggests MERS and SARS-like coronaviruses may not be the only bat coronaviruses that can cause disease outbreaks in humans, and further highlights the need for ongoing investigation of bats and their coronaviruses.
References
Anthony S, Ojeda-Flores R, Rico-Chávez O, Navarrete-Macias I, Zambrana-Torrelio C, Rostal M, et al. (2013). Coronaviruses in bats from Mexico. Journal of General Virology 94:1028-1038.
Applied Biosystems (2002) BigDye® Terminator v3.1 Cycle Sequencing Kit http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_081527.pdf. Accessed 15 October 2015
Breed AC, Field HE, Smith CS, Edmonston J, Meers J (2010). Bats Without Borders: Long-Distance Movements and Implications for Disease Risk Management. Ecohealth 7:204-212.
Brown LD, Cat TT, DasGupta A (2001). Interval Estimation for a proportion. Statistical Science 16:101-133.
Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, et al. (2006). Coronaviruses in bent-winged bats (Miniopterus spp.). Journal of General Virology 87:2461-2466.
Churchill S (2008). Australian bats, 2nd edition. Allen & Unwin, Crows Nest, New South Wales, Australia.
Cui J, Han N, Streicker D, Li G, Tang X, Shi Z, et al. (2007). Evolutionary relationships between bat coronaviruses and their hosts. Emerging Infectious Diseases 13:1526-1532.
Dominguez SR, OShea TJ, Oko LM, Holmes KV (2007). Detection of group 1 coronaviruses in bats in North America. Emerging Infectious Diseases 13:1295-300. DOI:10.3201/eid1309.070491.
Drexler JF, Corman VM, Drosten C (2014). Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Research 101:45-56.
Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. (2010). Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. Journal of Virology 84:11336-11349.
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine 348:1967-1976.
Ge X, Li J, Yang X, Chmura A, Zhu G, Epstein J, et al. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535-538.
Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, et al. (2008). Detection and Prevalence Patterns of Group I Coronaviruses in Bats, Northern Germany. Emerging Infectious Diseases 14:626-631.
Hutcheon JM, and Kirsch JAW (2006). A moveable face: deconstructing the Microchiroptera and a new classification of extant bats. Acta Chiropterologica 8:1-10.
International Committee on Taxonomy of Viruses (2011) Master species list #26 http://www.ictvonline.org/virusTaxonomy.asp?version=2011. Accessed 15 October 2015
Lau SK, Che XY, Woo PC, Wong BH, Cheng VC, Woo GK, et al. (2005a). SARS coronavirus detection methods. Emerging Infectious Diseases 11:1108-1111.
Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. (2005b). Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proceedings of the National Academy of Science 102:14040-14045.
Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, et al. (2007). Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 367:428-439.
Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB (2013). Detection of Coronaviruses in Bats of Various Species in Italy. Viruses 5:2679-2689.
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. (2005). Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676-679.
Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. (2013). Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerging Infectious Diseases 19:1819-1823.
Nei M, Kumar S (2000). Molecular evolution and phylogenetics. Oxford University Press, New York.
Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, et al. (2009). Distant Relatives of Severe Acute Respiratory Syndrome Coronavirus and Close Relatives of Human Coronavirus 229E in Bats, Ghana. Emerging Infectious Diseases 15:1377-1384.
Poon LL, Chan KH, Peiris JS (2004). Crouching tiger, hidden dragon: the laboratory diagnosis of severe acute respiratory syndrome. Clinical Infectious Diseases 38:297-299.
Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, et al. (2005). Identification of a novel coronavirus in bats. Journal of Virology 79:2001-2009.
Smith CS, de Jong CE, Field HE (2010). Sampling small quantities of blood from microbats. Acta Chiropterologica 12(1):255–258.
Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. (2010) Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329:676-9. DOI:10.1126/science.1188836. [Online August 6, 2010].
Streicker DG, Lemey P, Velasco-Villa A, Rupprecht CE. (2012) Rates of Viral Evolution Are Linked to Host Geography in Bat Rabies. PLoS Pathogens. 8: e1002720. DOI:10.1371/journal.ppat.1002720 [Online May 17, 2012].
Suzuki J, Sato R, Kobayashi T, Aoi T, Harasawa R (2014). Group B betacoronavirus in rhinolophid bats, Japan. Journal of Veterinary Medical Science 76:1267-1269.
Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30:2725-2729.
Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, et al. (2006). Prevalence and genetic diversity of coronaviruses in bats from China. Journal of Virology 80:7481-7490.
Tsuda S, Watanabe S, Masangkay J, Mizutani T, Alviola P, Ueda N, et al. (2012). Genomic and serological detection of bat coronavirus from bats in the Philippines. Archives of Virology 157:2349-2355.
Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y (2007). Evolutionary insights into the ecology of coronaviruses. Journal of Virology 81:4012-4020.
Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, et al. (2006). Molecular diversity of coronaviruses in bats. Virology 351:180-187.
Yu M, Stevens V, Berry JD, Crameri G, McEachern J, Tu C, et al. (2008) Determination and application of immunodominant regions of SARS coronavirus spike and nucleocapsid proteins recognized by sera from different animal species. Journal of Immunological Methods 331:1-12. DOI:10.1016/j.jim.2007.11.009 [Online December 17, 2007].
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, and Fouchier RAM (2012). Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. New England Journal of Medicine 367:1814-1820.
Acknowledgments
We thank the following colleagues for assistance with fieldwork: Andrew Breed and Amy Burroughs (then University of Queensland); Alan and Stacey Franks (Hollow Log Homes); Tim Kerlin (then Australian Quarantine and Inspection Service); Carol Palmer, John Burke, Chris Kinnaird and Damian Milne (then Northern Territory Parks and Wildlife Commission); Raina Plowright (then University of California, Davis); Jennifer McRobbie (then Cummings Tufts School of Veterinary Medicine); Anja Divljan (then University of Sydney) and Ximena Tolosa (then Biosecurity Queensland). For laboratory assistance, we thank Barry Rodwell, Bruce Corney and Ibrahim Diallo (Queensland Government Biosecurity Sciences Laboratory) and Gary Crameri, Meng Yu, Jennifer Barr, Mary Tachedjian and Dieter Bulach (CSIRO Australian Animal Health Laboratory). CS is particularly thankful to Dr Les Hall (then University of Queensland) who shared his knowledge of bats and spelunking. This study was supported by the Australian Biosecurity Cooperative Research Centre for Emerging Infectious Diseases. Additional support was provided by the Queensland Government Department of Agriculture and Fisheries. L-FW is supported in part by a National Research Foundation grant (NRF2012NRF-CRP001-056) in Singapore.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Smith, C.S., de Jong, C.E., Meers, J. et al. Coronavirus Infection and Diversity in Bats in the Australasian Region. EcoHealth 13, 72–82 (2016). https://doi.org/10.1007/s10393-016-1116-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10393-016-1116-x