
Abstract
Purpose
Autosomal dominant foveal hypoplasia (FVH1) is a rare disorder associated with mutations in the PAX6 gene. As an isolated disease entity, FVH1 does not include ocular disorders such as aniridia, microphthalmia, albinism, and achromatopsia. However, it only includes isolated foveal hypoplasia and foveal hypoplasia with presenile cataract. The purpose of this report is to present our findings in four patients from two families with FVH1 without visible ophthalmic macular abnormalities.
Study design
A review of the medical records of two families with FVH1 and genetic confirmation of mutations in the PAX6 gene.
Methods
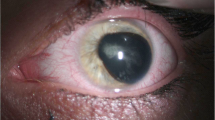
Fundus photographs, optical coherence tomographic (OCT) and OCT angiographic (OCTA) images, and slit-lamp anterior segment findings were determined. The type of mutation of the PAX6 gene was determined.
Results
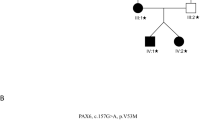
A 3-year-old girl (Patient 1) had signs and symptoms of an impairment in the development of vision without other retinal abnormalities OU. OCT images showed a shallow foveal pit, and OCTA showed the absence of the foveal avascular zone. The second patient (Patient 2) was a 6-year-old girl with unilateral mild cataract and shallow foveal pits OU. Similar shallow foveal pits were found in her asymptomatic mother (Patient 3) and maternal grandfather (Patient 4). Although the iris and posterior fundus were normal, all patients with FVH1 had goniodysgenesis. Genetic testing of the PAX6 gene revealed that Patient 1 had a novel heterozygous mutation (p.Asn365Lys) as a de novo mutation, and Patients 2, 3 and 4 had a novel heterozygous mutation (p.Pro20Ser).
Conclusions
Heterozygous mutations in the PAX6 gene can cause FVH1 with nearly normal appearing macula. FVH1 is difficult to diagnose, but detailed observations of the foveal structure and vasculature, and detecting the presence of goniodysgenesis can be helpful in identifying patients with FVH1.



Similar content being viewed by others

References
Mann I. Developmental abnormalities of the eye. London: BMA House; 1957. p. 141–143.
Duke-Elder S. System of ophthalmology. London: Mosby; 1963. p. 652–657.
Harrison R, Hoefnagel D, Hayward JN. Congenital total color blindness: a clincopathological report. Arch Ophthalmol. 1960;64:685–92.
Curran RE, Robb RM. Isolated foveal hypoplasia. Arch Ophthalmol. 1976;94:48–50.
Oliver MD, Dotan SA, Chemke J, Abraham FA. Isolated foveal hypoplasia. Br J Ophthalmol. 1987;71:926–30.
O'Donnell FE Jr, Pappas HR. Autosomal dominant foveal hypoplasia and presenile cataracts. A new syndrome. Arch Ophthalmol. 1982;100:279–81.
Poulter JA, Al-Araimi M, Conte I, van Genderen MM, Sheridan E, Carr IM, et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93:1143–50.
Perez Y, Gradstein L, Flusser H, Markus B, Cohen I, Langer Y, et al. Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet. 2014;22:703–6.
Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M. PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet. 1996;13:141–2.
Hanson I, Churchill A, Love J, Axton R, Moore T, Clarke M, et al. Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet. 1999;8:165–72.
Rufai SR, Thomas MG, Purohit R, Bunce C, Lee H, Proudlock FA, et al. Can structural grading of foveal hypoplasia predict future vision in infantile nystagmus?: A longitudinal study. Ophthalmology. 2020;127:492–500.
Wilk MA, McAllister JT, Cooper RF, Dubis AM, Patitucci TN, Summerfelt P, et al. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014;55:4186–98.
Hingorani M, Williamson KA, Moore AT, van Heyningen V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 2009;50:2581–90.
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–D268268.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Springer AD, Hendrickson AE. Development of the primate area of high acuity. 1. Use of finite element analysis models to identify mechanical variables affecting pit formation. Vis Neurosci. 2004;21:53–62.
Sato R, Kunikata H, Asano T, Aizawa N, Kiyota N, Shiga Y, et al. Quantitative analysis of the macula with optical coherence tomography angiography in normal Japanese subjects: the Taiwa Study. Sci Rep. 2019;9:8875.
Marmor MF, Choi SS, Zawadzki RJ, Werner JS. Visual insignificance of the foveal pit: reassessment of foveal hypoplasia as fovea plana. Arch Ophthalmol. 2008;126:907–13.
Matsushita I, Nagata T, Hayashi T, Kimoto K, Kubota T, Ohji M, et al. Foveal hypoplasia in patients with Stickler syndrome. Ophthalmology. 2017;124:896–902.
Recchia FM, Recchia CC. Foveal dysplasia evident by optical coherence tomography in patients with a history of retinopathy of prematurity. Retina. 2007;27:1221–6.
Hanson IM. PAX6 and congenital eye malformations. Pediatr Res. 2003;54:791–6.
Thomas S, Thomas MG, Andrews C, Chan WM, Proudlock FA, McLean RJ, et al. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet. 2014;22:344–9.
Chauhan BK, Medsinge A, Baumgartner MP, Scanga HL, Kamakari S, Gajdosova E, et al. Case series: pyramidal cataracts, intact irides and nystagmus from three novel PAX6 mutations. Am J Ophthalmol Case Rep. 2018;10:172–9.
Mirrahimi M, Sabbaghi H, Ahmadieh H, Jahanmard M, Hassanpour K, Suri F. A novel PAX6 mutation causes congenital aniridia with or without retinal detachment. Ophthalmic Genet. 2019;40:146–9.
Aggarwal S, Jinda W, Limwongse C, Atchaneeyasakul LO, Phadke SR. Run-on mutation in the PAX6 gene and chorioretinal degeneration in autosomal dominant aniridia. Mol Vis. 2011;17:1305–9.
Acknowledgements
We thank the patients for granting permission to publish this study. We also thank Duco Hamasaki, Professor Emeritus, Bascom Palmer Eye Institute, University of Miami, Miami, Florida, for his critical comments and valuable assistance.
Funding
This study was supported by Grants-in-Aid for Scientific Research C (HK, 20K09818) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
I. Matsushita, None; H. Morita, None; H. Kondo, Lecture fee (Santen, Bayer, Kowa, Senju, Otsuka, RE Medical), Grant, Lecture fee (Alcon, AMO Japan, Novartis).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Corresponding Author: Hiroyuki Kondo
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Matsushita, I., Morita, H. & Kondo, H. Autosomal dominant foveal hypoplasia without visible macular abnormalities and PAX6 mutations. Jpn J Ophthalmol 64, 635–641 (2020). https://doi.org/10.1007/s10384-020-00766-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10384-020-00766-9