
Abstract
Following severe population decline and local extinction due to massive habitat destruction and persecution, wildcats have recently reappeared in several parts of Germany’s low mountain region. It remains unknown how this reemergence occurred, specifically if local populations have been overlooked at low densities or if the species has successfully spread across the highly fragmented anthropogenic landscape. In the central German Rhön Mountains, for instance, wildcats were believed to be extinct during most of the twentieth century, however, the species was recently detected and subsequent genetic monitoring found the presence of a sizeable population. In this study, we used microsatellite and SNP genotypes from 146 wildcat individuals from 2008 to 2017 across a ~ 15,000 km2 area in the central German low mountain region to understand the population re-establishment of wildcats in the region. Bayesian clustering and subsequent analyses revealed that animals in the Rhön Mountains appear to be a mix from the two adjacent populations in the North and South of the area, suggesting a recent range expansion from two different directions. Both populations meet in the Rhön Biosphere Reserve, leading to an admixture of the northern, autochthonous, and the southern reintroduced wildcat population. While we cannot completely exclude the possibility of undetected population persistence, the high genetic homogeneity in the central German wildcat population and the lack of any signatures of past population decline in the Rhön favor a scenario of natural expansion. Our findings thus suggest that wildcats are well capable of rapid range expansion across richly structured landscape mosaics consisting of open land, settlements, and forest patches and document the potential of massive non-invasive genetic sampling when aiming to reconstruct the complex population and range dynamics of wildlife.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many populations of large- and medium-sized carnivores are currently re-expanding their ranges across the densely populated and anthropogenically modified landscapes of central Europe (Chapron et al. 2014). These altered landscapes exhibit high levels of habitat fragmentation, which is the disruption of continuous stretches of suitable habitat (Schadt et al. 2002). Habitat fragmentation can negatively affect the viability of populations and species. However, the extent to which species are affected varies significantly (Haddad et al. 2015). A species’ reaction to cultural landscapes depends heavily on the size of suitable habitat, the distance between habitat patches, and the individual species’ life history traits (Caruso et al. 2016). Carnivores, for instance, are at a heightened risk of being negatively impacted in fragmented landscapes due to their relatively large home ranges and low population densities (Crooks 2002; Noss et al. 1996; Woodroffe 1998). Therefore, investigating how carnivores successfully disperse and establish viable populations in altered landscapes is important to understand the potential for long-term existence of wildlife in human-dominated landscapes (Riley et al. 2003).
The European wildcat is a flagship species for nature conservation in Germany, as it primarily relies on highly structured natural deciduous and mixed forests (Driscoll and Nowell 2010). This reliance on connected forests has made it a pillar for the preservation of natural habitats. Recently, the wildcat has made a reemergence across the central German low mountain region (Steyer et al. 2016), providing a relevant example to understand dispersal dynamics in a highly fragmented landscape.
Historically, European wildcat populations were heavily depleted due to human persecution until the early twentieth century. In Germany, the species was restricted to few refugial areas within the low mountain region, such as the Pfälzerwald, Eifel, and the Harz Mountains (Eckert et al. 2010). After hunting pressure was reduced, distance between refugial areas, habitat fragmentation, and increasing anthropogenic development left local wildcat populations geographically isolated (Hille et al. 2000; Pierpaoli et al. 2003). Considering the possible negative effects of isolated populations, including inbreeding and genetic drift, efforts to connect these distinct wildcat populations have become a conservation priority (Klar et al. 2012; Mattucci et al. 2016). Given the concern for wildcat population connectivity, genetic monitoring relying on hair trapping was established across known wildcat territories (Klar et al. 2008; Simon and Hupe 2008; Steyer et al. 2013). The results of increased monitoring showed strong evidence for the recent recovery of wildcat populations in various areas (Steyer et al. 2013; Steyer et al. 2016). Most recently, the species had been documented in areas distant from the known source regions, such as in the Bayerischer Wald in the Southeast and the Lüneburger Heide in Northern Germany (unpublished data). These new detections of the species in areas outside long-standing refugia and reintroduction sites suggest that the wildcat is capable of rapid dispersal and establishment across anthropogenically modified landscapes (scenario 1). However, it remains unknown if the species may have persisted in small, overlooked populations that are now increasing locally (scenario 2). Revealing the origin of wildcat reemergence is of considerable importance, especially considering the first scenario, expansion from refugia and reintroduction areas, which would imply that effective dispersal and population establishment of mobile mammals such as wildcats may be less obstructed by habitat fragmentation and barriers within anthropogenic landscapes than previously assumed (Klar et al. 2008; Jerosch et al. 2018; Balkenhol and Waits 2009).
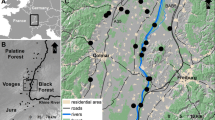
Here, we aim to disentangle the above-mentioned recolonization scenarios by investigating the fine-scale genetic population structure of wildcats in a region where the species was recently detected, namely the Rhön Biosphere Reserve (RBR). The RBR is located on the southeastern edge of published wildcat distribution in the central low mountain region (Fig. 1) (Klar et al. 2009). The RBR is a biodiversity-rich landscape, dominated by meadows and open land, surrounded by multiple large highways (A7, A71, A4) (Jedicke 2013). The RBR is home to a variety of protected species, including the wildcat. Wildcats in the RBR were thought to be extinct during most of the twentieth century until first genetic detections of wildcats occurred between 2007 and 2009 (Birlenbach et al. 2009). Beginning in 2009, under the frame of various monitoring projects (Table S1), the RBR and surroundings were heavily searched for evidence of wildcat, resulting in the detection of a substantial presence of wildcat in this region (Reiners et al. 2014; Thein 2008). Scenario 1 would imply wildcats expanded from adjacent populations into the RBR. Given the regional history, there are two possible source populations; (i) the refugial population in the north and (ii) the reintroduced population in the adjacent Spessart Mountains (Fig. 1). The known wildcat distribution to the north of the RBR was documented since before 2009 as a known source of a stable wildcat population (Birlenbach et al. 2009). The wildcat reintroduction in the Spessart region was launched in 1984 under the frame of a long-term reintroduction project. Approximately 600 wildcats were released until 2008 (Büttner and Worel 1990). Additionally, in 2005, six wildcats were reintroduced in the Neuwirthauser Forest, which is at the southernmost part of the RBR. It remains unknown if this reintroduction was successful and contributed to the reemergence in the RBR or if the current population resembles dispersal of individuals from the north. Scenario 2 would indicate a local increase from a small, overlooked relict population within the Rhön Mountains, which would likely result in population sub-structuring, due to the loss of rare alleles in small isolated populations, indicating the presence that a relict population survived and is now expanding in the low mountain region (Excoffier et al. 2009).

Map of 146 wildcat samples used for combined SNP and microsatellite approach (blue) and 439 wildcat samples analyzed only with msats (orange) from 2004 to 2017 used for analysis showing the known wildcat distribution (KWD), Rhön Biosphere Reserve (RBR), and Spessart reintroduction (SPR) as well as the forested area based on ATKIS (FOR). Map of the study area is shown in the red box within the inlet
The present study aims to disentangle the two proposed scenarios to shed light on the origin of wildcats in this low mountain region. For this, data was obtained from a large-scale genetic monitoring program conducted over the past 10 years in Germany. From this monitoring program, we genotyped a subset of samples with SNP and microsatellite markers to reveal the genetic makeup in the RBR. We compared the genetic structure of wildcats in the RBR with both adjacent source and reintroduced populations (Steyer et al. 2016) to test the two scenarios explaining the emergence of the species in the RBR given above.
Materials and methods
Study site
The study area comprised the Rhön Mountains, including the RBR and surroundings (Fig. 1). The reserve spans over 2433 km2, which was expanded in 2014 to include the southern Rhön with the NF. The Rhön is a low mountain range ranging from 250 to 950 m, with approximately 40% forested land. Within the study area, the closest potential source populations were included; mainly the region surrounding the National Park Hainich in the north, and the Spessart Mountains, bordering the Rhön Mountains to the southwest. The Hainich Mountains range from 225 to 494 m and are the most expansive broad-leaved forest, the primary habitat of the wildcat, in Germany. The Spessart Mountains include one of Germany’s most forested areas, peaking at 586 m. In total, our study consisted of approximately 15,000 km2 of variable habitats.
Sample collection and laboratory methodologies
Multiple opportunistic and standardized monitoring projects collected samples from 2004 to 2017. Opportunistic wildcat roadkill samples were collected throughout the study period. In addition, multiple monitoring projects began in 2008, most notably Rhoen Natur e.V., BUND Wildkatzensprung, and Biosphärenreservat Rhön projects, which resulted in intense monitoring within the RBR during this time (for a complete list, see supplementary Table S1). In this study, we combined microsatellite and SNP genotyping methods. We took 119 wildcat microsatellite genotypes from 2004 to 2013 from a German-wide study on wildcat population structure (Steyer et al. 2016) and added 49 additional genotypes from 2014 to 2017 to be further analyzed using a wildcat specific SNPtype™ marker panel (von Thaden et al. 2020). Microsatellite genotypes were considered for SNP genotyping if they had a minimum of 11 loci and showed amplification success rates of 80%. Additionally, this selection excluded potential hybrids as there are low rates of hybridization across the study area (Steyer et al. 2018). The samples were then selected to create an even distribution in location, time, and sample type. Of the selected genotypes, 64 samples originated from tissue of dead found wildcats, and the remaining 104 samples are hair samples from various monitoring projects.
We used the primers LF4 (Eckert et al. 2010) and H16498 (Kocher et al. 1989) to sequence a 110 bp fragment of the mitochondrial control region following the protocol in Steyer et al. (2016). All samples were analyzed using 14 microsatellite markers and a zink finger sex marker (Hartmann et al. 2013). For non-invasively collected samples, the multiple tube approach using three replicates per sample was applied (Hartmann et al. 2013). Individualization was carried out through a custom R script and duplicated individuals were removed before further analysis (see Steyer et al. 2016 for further details). The microsatellite genotypes utilized from the Steyer et al. (2016) study were compared with the samples collected between 2014 and 2017 to look for batch effects. As none was found, the sample sets were combined.
The SNP genotyping of wildcat samples (n = 168) was performed on a EP1 platform (Fluidigm Corp., USA) using microfluidic 96.96 Dynamic Arrays™. Detailed methods and wildcat-specific SNPtype™ Assays are presented in von Thaden et al. (2017) and von Thaden et al. (2020). The SNPtype™ marker panel encompasses 84 loci selected for individual and population identification, 10 loci for hybrid detection, and 2 SRY-linked loci. All SNP experiments included four no template controls (NTC) per array and non-invasively collected hair samples were triplicated to detect potential errors. From 168 samples, seven samples did not show sufficient (> 70%) amplification success to be scored and used in downstream analysis. Subsequent individualization resulted in 146 individuals to be analyzed using the combined SNP and microsatellite data.
Data analyses
First, we tested the combined SNP and microsatellite data set for isolation by distance (IBD) effects. We performed an IBD analysis as implemented in GeneAlEx 6.5 (Peakall and Smouse 2012) to account for genetic differentiation solely based on geographical position of individuals. The program STRUCTURE v2.3.4 (Pritchard et al. 2000) was used to evaluate population genetic structure. After 100,000 steps of burn-in, 200,000 MCMC steps were performed with admixture model and correlated allele frequencies using a range of K 1–10 with 10 iterations. We used Structure Harvester (Earl and vonHoldt 2012) with the Evanno method (Evanno et al. 2005) to determine the most likely number of population clusters. The replicates were consolidated with the software CLUMPP (Jakobsson and Rosenberg 2007) using the GREEDY algorithm.
Further review of the spatial structure was carried out with the ADEGENET package in R (v.3.4.2) using a spatial principal component analysis (sPCA) (Jombart 2008). No requirements of the data to meet Hardy-Weinberg expectations or linkage equilibrium are needed for this method. In addition to the genetic data, sPCA also uses spatial information and is particularly suitable for the analysis of weak genetic structures (Storfer et al. 2007). sPCA relies on Moran’s I (Moran 1948, 1950) to identify spatial patterns within the genetic structure of the sampled individuals. The method distinguishes between global scores, which indicate gradients in allele frequencies, and local scores, indicating differences in neighboring samples.
Standard genetic diversity indices including expected and observed heterozygosity, allelic richness, and population pairwise FST values were calculated for the combined SNP and microsatellite genotypes using Arlequin version 3.5.2.2 (Excoffier and Lischer 2010).
Results
Of the 146 genotyped individuals, mtDNA haplotypes were successfully determined in 134 individuals and corresponded to SNG-HP-FS03/-04/-06/-22/-23 (Steyer et al. 2016). Haplotype SNG-HP-FS23 was found in eight individuals solely in the southern part of the study region (Fig. 2b). The other four haplotypes appeared in all parts of the study area.

Map showing the spatial genetic structure as assessed by sPCA analysis and haplotype map. a Individual scores from the first principal component: large blue squares indicated a highly positive score, large red squares indicated highly negative scores. b Map of individuals within the study area with the haplotype 23 (red) and all other haplotypes (3, 4, 6, 22)
Analysis of genetic structure based on combined SNP and microsatellite genotypes revealed no evidence of isolation by distance (IBD; Mantel test: r = 0.011, p = 0.48). Bayesian clustering implemented in STRUCTURE indicated K = 2 as the most likely number of clusters within the study area. Separation of these clusters could not be attributed to a geographical pattern, with individuals clearly showing representation from both clusters (Fig. 3).

STRUCTURE results from K = 2, which was suggested by Structure Harvester as the most likely value from K 1 to 10. Each bar represents a single individual. The coloration corresponds to the estimated proportions of posterior probability assignments of each sample to each cluster
Clustering with sPCA resulted in a significant global structure, indicating correlation between the genetic and geographic distances (p = 0.01). However, no significant local structure was found (p = 0.14). A plot of lagged scores from the first principal component suggested the global structure is linked to a north-south genetic border, with a transitional area within the RBR (Fig. 2a). Subsequent principal components show weaker genetic structure although the eigenvalues suggest the first principal component explains most of the genetic variance found in wildcat individuals from the study area (Fig. S1).
Standard measures of genetic diversity were carried out based on the two clusters defined by the sPCA (Table 1). When looking at two clusters, observed (0.41, 0.43) and expected (0.42, 0.43) heterozygosity values from the north and south, respectively, were highly similar. Allelic richness was also identical (2.45, 2.45) along with a comparable fixation index (0.047, 0.011). The RBR, which comprised much of the south cluster, showed no local genetic signature such as higher allelic richness or specific clustering in the region. We also carried out pairwise FST analysis based on the two clusters, which were not significantly differentiated from each other (Table 2). We also compared our dataset with a larger, microsatellite-only sample set of 439 individuals originating from a previous study (Steyer et al. 2016), which revealed some sub-structuring in the south, specifically between the reintroduced region and the RBR (Fig. S2).
Discussion
In recent decades, wildcat populations across low mountain regions in Germany showed signs of expansion, despite a considerably fragmented landscape (Hartmann et al. 2013; Würstlin et al. 2016). While the recolonization process has been well documented (Canters et al. 2005; Nussberger et al. 2018; Streif et al. 2017), few attempts have been made to distinguish between active range expansion and locally growing populations. We assessed regional population structure of wildcat based on available samples from various monitoring projects in the region (Table S1). We investigated population sub-structuring and fine-scale genetic diversity to shed light on two possible scenarios for the rapid appearance of wildcats within this low mountain region.
The number of wildcat individuals found within the study region between 2009 and 2014 (Fig. 1) indicates the presence of a viable wildcat population within the RBR. In part, this high number of wildcat detections can be attributed to the intense monitoring activities by multiple concurrent projects (Table S1). However, it appears unlikely that the marked increase of wildcat evidence since 2009 can be explained solely by increased monitoring activities. To our knowledge, local experts have continuously looked for wildcat presence in the region, making it unlikely that a population remained undetected for decades in the Rhön (Franz Müller, pers. comm.). Roadkill monitoring has occurred since 2004 rather opportunistically, involving local authorities, conservationists, and hunters. In addition, the overall observed trend of recent wildcat expansion across various regions within Germany (Steyer et al. 2016; Streif et al. 2017; Würstlin et al. 2016) supports the observed pattern of population increase within the study area.
The past reintroduction of wildcats in the Spessart allows for tracking possible wildcat expansion in this region through a unique mtDNA haplotype (Steyer et al. 2016). While haplotypes SNG-HP-FS03/-04/-06/-22 are the most common haplotypes in German wildcat populations (Steyer et al. 2016), SNG-HP-FS23 is restricted to the Spessart Mountains (Fig. 2b). While this gives clear indication that reintroduced wildcats have successfully established in the Spessart region, the distribution of this haplotype also documents a lack of substantial spread into the RBR. Our findings of the haplotype SNG-HP-FS23 being largely restricted to the Spessart reintroduction area suggests that the expansion into the Rhön area is mainly driven by male dispersal from the Southern reintroduction area and confirms the general observation of male-dominated dispersal in carnivores (Støen et al. 2005).
Our results from population structure analysis indicate a highly admixed population that comprises the northern refugial and the reintroduced populations. The sPCA results indicate that wildcats in the northern area of the RBR are genetically indistinguishable from northern refugia, which derive from the central German wildcat population described in Steyer et al. (2016) (Fig. 2a). In addition, the data suggests that wildcats from the reintroduction are moving into the RBR. Interestingly, STRUCTURE results show a highly admixed population with no clear geographic boundaries (Fig. 3). This may be explained by overall weak sub-structuring (FST < 0.05) (Hubisz et al. 2009; Stift et al. 2019). The FST measures also showed no significant deviation between the northern and southern clusters defined by the sPCA. This hints at a highly admixed population throughout the study region.
We found no evidence of an overlooked population within the RBR, as no private alleles were discovered in the area and we did not find any genetic sub-structure separating the Rhön from adjacent regions. Thus, the most probable explanation for the observed genetic pattern is a recolonization from both (northern and southern) directions. In case of overall low sub-structure within the central German wildcat population, which has been found previously (Steyer et al. 2016) and is confirmed in this study, a scenario of local population size increase together with significant dispersal from adjacent areas appears feasible. Still, the lack of evidence for wildcat appearance prior to 2008 makes the first option more likely and both scenarios ultimately require substantial dispersal from adjacent regions, suggesting a significant permeability of the landscape for wildcats.
Our results imply that wildcats can disperse through human-dominated landscapes. Land use within the newly recolonized RBR comprises 41% forested land (Jedicke 2013), with the remaining being settlement, open meadows, or arable land. Our study suggests that wildcats can, within a few years or few generations, recolonize a region with a substantial proportion of open land. This reemergence implies the wildcat is not significantly isolated due to road infrastructure, and can establish home ranges in primarily agricultural habitats, which has been supported by recent studies showing similar results (Jerosch et al. 2017; Jerosch et al. 2018; Klar et al. 2009; Würstlin et al. 2016). The presence of green bridges, built in 2011, potentially facilitates the exchange between individuals on either side of the A7 highway, as has been shown in other regions (Pir et al. 2011). The main factor determining habitat choice in wildcats is thought to be distance to forest (Sarmento et al. 2006). Jerosch et al. (2018) highlight structural heterogeneity in open landscapes as a determining factor allowing wildcats to persist in human-dominated landscapes. Therefore, the presence of rich-structured mosaics of open land and forest patches might have provided suitable conditions for successful recolonization in the RBR.
Our findings confirm a recolonization process of the wildcat in the Rhön Mountains, which has important implications for current wildcat conservation strategies. The results mainly suggest that connecting suitable habitat through stepping stones of rich-structured patches, rather than continuous forest, may be sufficient for wildcat dispersal. Therefore, it is important to conserve landscapes of rich-structured mosaics of open land and forest patches, similar to the Rhön, as habitat fragmentation continues to occur. As a flagship species, efforts to create heterogeneous landscapes will help other forest-dwelling species to migrate between patches.
References
Balkenhol N, Waits LP (2009) Molecular road ecology: exploring the potential of genetics for investigating transportation impacts on wildlife. Mol Ecol 18(20):4151–4164. https://doi.org/10.1111/j.1365-294X.2009.04322.x
Birlenbach K, Klar N, Jedicke E, Wendel M, Wachendörfer V, Fremuth W, TAM K, Mölich T, Vogel B (2009) Aktionsplan zum Schutz der Europäischen Wildkatze in Deutschland: Schutzkonzept für eine Zielart des überregionalen Waldbiotopverbunds. Naturschutz und Landschaftplanung 41
Büttner K, Worel G (1990) Wiedereinbürgerung der Europäischen Wildkatze in Bayern – in Projekt des Bundes Naturschutz in Bayern. Waldhygiene 18:169–176
Canters KJ, Thissen JBM, van Diepenbeck A, Jansman HAH, Goutbeck K (2005) The wildcat (Felis silvestris) finally recorded in the Netherlands. Lutra 48:67–90
Caruso N, Lucherini M, Fortin D, Casanave EB (2016) Species-specific responses of carnivores to human-induced landscape changes in central Argentina. PLoS One 11:e0150488. https://doi.org/10.1371/journal.pone.0150488
Chapron G, Kaczensky P, Linnell JDC, von Arx M, Huber D, Andrén H, López-Bao JV, Adamec M, Álvares F, Anders O, Balčiauskas L, Balys V, Bedő P, Bego F, Blanco JC, Breitenmoser U, Brøseth H, Bufka L, Bunikyte R, Ciucci P, Dutsov A, Engleder T, Fuxjäger C, Groff C, Holmala K, Hoxha B, Iliopoulos Y, Ionescu O, Jeremić J, Jerina K, Kluth G, Knauer F, Kojola I, Kos I, Krofel M, Kubala J, Kunovac S, Kusak J, Kutal M, Liberg O, Majić A, Männil P, Manz R, Marboutin E, Marucco F, Melovski D, Mersini K, Mertzanis Y, Mysłajek RW, Nowak S, Odden J, Ozolins J, Palomero G, Paunović M, Persson J, Potočnik H, Quenette P-Y, Rauer G, Reinhardt I, Rigg R, Ryser A, Salvatori V, Skrbinšek T, Stojanov A, Swenson JE, Szemethy L, Trajçe A, Tsingarska-Sedefcheva E, Váňa M, Veeroja R, Wabakken P, Wölfl M, Wölfl S, Zimmermann F, Zlatanova D, Boitani L (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519. https://doi.org/10.1126/science.1257553
Crooks KR (2002) Relative sensitivities of mammalian carnivores to habitat fragmentation. Conserv Biol 16:488–502. https://doi.org/10.1046/j.1523-1739.2002.00386.x
Driscoll CA, Nowell K (2010) Felis silvestris. In: IUCN red list of threatened species. International union for conservation of nature. http://www.iucnredlist.org/
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eckert I, Suchentrunk F, Markov G, Hartl GB (2010) Genetic diversity and integrity of German wildcat (Felis silvestris) populations as revealed by microsatellites, allozymes, and mitochondrial DNA sequences. Mamm Biol 75:160–174. https://doi.org/10.1016/j.mambio.2009.07.005
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501. https://doi.org/10.1146/annurev.ecolsys.39.110707.173414
Haddad NM, Brudvig LA, Clobert J, Davies KF, Gonzalez A, Holt RD, Lovejoy TE, Sexton JO, Austin MP, Collins CD, Cook WM, Damschen EI, Ewers RM, Foster BL, Jenkins CN, King AJ, Laurance WF, Levey DJ, Margules CR, Melbourne BA, Nicholls AO, Orrock JL, Song D-X, Townshend JR (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv 1:e1500052. https://doi.org/10.1126/sciadv.1500052
Hartmann SA, Steyer K, Kraus RHS, Segelbacher G, Nowak C (2013) Potential barriers to gene flow in the endangered European wildcat (Felis silvestris). Conserv Genet 14:413–426. https://doi.org/10.1007/s10592-013-0468-9
Hille A, Pelz O, Trinzen M, Schlegel M, Peters G (2000) Using microsatellite markers for genetic individualization of European wildcats (Felis silvestris) and domestic cats. Bonn zool Beitr 49:165–176
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9(5):1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jedicke E (2013) Report for the periodic review of Rhön UNESCO Biosphere Reserve. Biosphärenreservat Rhön Verwaltung Thüringen. http://www.unesco.org/new/fileadmin/MULTIMEDIA/HQ/SC/pdf/Periodic_Review_Rhon_2013.pdf
Jerosch S, Götz M, Roth M (2017) Spatial organisation of European wildcats (Felis silvestris silvestris) in an agriculturally dominated landscape in Central Europe. Mammalian Biology - Zeitschrift für Säugetierkunde 82:8–16. https://doi.org/10.1016/j.mambio.2016.10.003
Jerosch S, Kramer-Schadt S, Götz M, Roth M (2018) The importance of small-scale structures in an agriculturally dominated landscape for the European wildcat (Felis silvestris silvestris) in central Europe and implications for its conservation. J Nat Conserv 41:88–96. https://doi.org/10.1016/j.jnc.2017.11.008
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Klar N, Fernández N, Kramer-Schadt S, Herrmann M, Trinzen M, Büttner I, Niemitz C (2008) Habitat selection models for European wildcat conservation. Biol Conserv 141:308–319. https://doi.org/10.1016/j.biocon.2007.10.004
Klar N, Herrmann M, Kramer-Schadt S (2009) Effects and mitigation of road impacts on individual movement behavior of wildcats. J Wildl Manag 73:631–638. https://doi.org/10.2193/2007-574
Klar N, Herrmann M, Henning-Hahn M, Pott-Dörfer B, Hofer H, Kramer-Schadt S (2012) Between ecological theory and planning practice: (re-)connection of forest patches for the wildcat in Lower Saxony. Biol Conserv 105:376–384
Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. PNAS 86:6196–6200
Mattucci F, Oliveira R, Lyons LA, Alves PC, Randi E (2016) European wildcat populations are subdivided into five main biogeographic groups: consequences of Pleistocene climate changes or recent anthropogenic fragmentation? Ecol Evol 6:3–22. https://doi.org/10.1002/ece3.1815
Moran PAP (1948) The interpretation of statisical maps. J R Stat Soc B 10:243–251
Moran PAP (1950) A test for the serial independence of residuals. Biometrika 37:178–181
Noss RF, Quigley HB, Hornocker MG, Merrill T, Paquet PC (1996) Conservation Biology and Carnivore Conservation in the rocky mountains. Conserv Biol 10:949–963. https://doi.org/10.1046/j.1523-1739.1996.10040949.x
Nussberger B, Currat M, Quilodran CS, Ponta N, Keller LF (2018) Range expansion as an explanation for introgression in European wildcats. Biol Conserv 218:49–56
Peakall R, Smouse PE (2012) GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pierpaoli M, Biro ZS, Herrmann M, Hupe K, Fernandes M, Ragni B, Szemethy L, Randi E (2003) Genetic distinction of wildcat (Felis silvestris) populations in Europe, and hybridization with domestic cats in Hungary. Mol Ecol 12:2585–2598. https://doi.org/10.1046/j.1365-294X.2003.01939.x
Pir JB, Schauls R, Dietz M, Simon O (2011) Bedeutung von Wildbrücken zur Vernetzung von Wanderkorridoren für die Europäische Wildkatze (Felis silvestris silvestris Schreber, 1777) am Beispiel von Pettingen/Mersch (Luxemburg). Bulletin de la SNL 112:59–72
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reiners TE, Tiesmeyer A, Carl S, Steyer K, Nowak C (2014) Wissenschaftliche Begleituntersuchung zum länderübergreifenden Wildkatzenmonitoring im Biosphärenreservat Rhön 2013-2014. Im Auftrag der Regierung von Unterfranken Bayerische Verwaltungsstelle Biosphärenreservat Rhön. https://www.biosphaerenreservat-rhoen.de/fileadmin/media/fotos/Projekte/Wildkatze/2014_wildkatze_genetik_schlussbericht.pdf
Riley SPD, Sauvajot RM, Fuller TK, York EC, Kamradt DA, Bromley C, Wayne RK (2003) Effects of urbanization and habitat fragmentation on bobcats and coyotes in Southern California. Conserv Biol 17:566–576. https://doi.org/10.1046/j.1523-1739.2003.01458.x
Sarmento P, Cruz J, Tarroso P, Fonseca C (2006) Space and habitat selection by female European wild cats (Felis silvestris silvestris). Wildl Biol Pract 2. https://doi.org/10.2461/wbp.2006.2.10
Schadt S, Revilla E, Wiegand T, Knauer F, Kaczensky P, Breiten Moser U, Bufka L, Cerveny J, Koubek P, Huber T, Stanisa C, Trepl L (2002) Assessing the suitability of central European landscapes for the reintroduction of Eurasian lynx. J Appl Ecol 39:189–203
Simon O, Hupe K (2008) Nachweis der Wildkatze (Felis silvestris silvestris) im Nationalpark Kellerwald-Edersee und den umgebenden Waldgebieten der Breiten Struth, des Hohen Kellers und des nördlichen Burgwaldes mit Hilfe der Lockstockmethode im Winter 2007/08. Jahrbuch Naturschutz in Hessen 12:18–25
Steyer K, Simon O, Kraus RHS, Haase P, Nowak C (2013) Hair trapping with valerian-treated lure sticks as a tool for genetic wildcat monitoring in low-density habitats. Eur J Wildl Res 59:39–46. https://doi.org/10.1007/s10344-012-0644-0
Steyer K, Kraus RHS, Mölich T, Anders O, Cocchiararo B, Frosch C, Geib A, Götz M, Herrmann M, Hupe K, Kohnen A, Krüger M, Müller F, Pir JB, Reiners TE, Roch S, Schade U, Schiefenhövel P, Siemund M, Simon O, Steeb S, Streif S, Streit B, Thein J, Tiesmeyer A, Trinzen M, Vogel B, Nowak C (2016) Large-scale genetic census of an elusive carnivore, the European wildcat (Felis s. silvestris). Conserv Genet 17:1183–1199. https://doi.org/10.1007/s10592-016-0853-2
Steyer K, Tiesmeyer A, Muñoz-Fuentes V, Nowak C (2018) Low rates of hybridization between European wildcats and domestic cats in a human-dominated landscape. Ecol Evol 8:2290–2304. https://doi.org/10.1002/ece3.3650
Stift M, Kolář F, Meirmans PG (2019) STRUCTURE is more robust than other clustering methods in simulated mixed-ploidy populations. Heredity 123(4):429–441. https://doi.org/10.1038/s41437-019-0247-6
Støen O-G, Bellemain E, Sæbø S, Swenson JE (2005) Kin-related spatial structure in brown bears Ursus arctos. Behav Ecol Sociobiol 59(2):191–197. https://doi.org/10.1007/s00265-005-0024-9
Storfer A, Muerphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape genetics. Heredity (Edinb) 98:128–142
Streif S, Kohnen A, Willhelm C (2017) The European wildcat in the Upper Rhine Valley and region Kaiserstuhl:16–18. Annual Report. Forest Research Institute Baden-Württemberg (FVA). https://www.fva-bw.de/fileadmin/publikationen/jahrber/jb20016_gb.pdf
Thein J (2008) Die Wildkatze in Bayern. Aktivitäten im Wildkatzenprojekt des Bundes Naturschutz im Jahr 2008. Unveröffentlichtes Gutachten im Auftrag des Bund Naturschutz, Nürnberg. 35 Seiten.
von Thaden A, Cocchiararo B, Jarausch A, Jüngling H, Karamanlidis AA, Tiesmeyer, A. … Muñoz-Fuentes, V. (2017) Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci Rep 7:83. https://doi.org/10.1038/s41598-017-10647-w
von Thaden A, Nowak C, Tiesmeyer A, Reiners TE, Alves PC, Lyons, L. A. … Cocchiararo, B. (2020) Applying genomic data in wildlife monitoring: development guidelines for genotyping degraded samples with reduced single nucleotide polymorphism (SNP) panels. Mol Ecol Resour 20:662–680. https://doi.org/10.1111/1755-0998.13136
Woodroffe R (1998) Edge effects and the extinction of populations inside protected areas. Science 280:2126–2128. https://doi.org/10.1126/science.280.5372.2126
Würstlin S, Segelbacher G, Streif S, Kohnen A (2016) Crossing the Rhine: a potential barrier to wildcat (Felis silvestris silvestris) movement? Conserv Genet 17:1435–1444. https://doi.org/10.1007/s10592-016-0874-x
Acknowledgments
We are deeply grateful to all volunteers who collected hair samples from lurestick traps across the study range. Special thanks go to Ulrike Schade, Doris Pokorny (Biosphärenreservat Rhön, Verwaltungsstelle Bayern), Eckard Jedicke and Matthias Metzger (Rhönnatur e.V.), Wolfgang Fremuth and Michael Brombacher (FZS), and Kai Frobel (BN). All DNA sequencing and fragment length analyses in this study were performed in the laboratory center of the Senckenberg BiK-F funded by the LOEWE initiative (Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz) of the Federal State of Hessen. We highly appreciate the laboratory assistance provided by Mascha Siemund, Susanne Carl, and Berardino Cocchiararo (Senckenberg).
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding for this project comes from the Frankfurt Zoological Society (FZS), the Allianz Umweltstiftung, the Biosphärenreservat Rhön, Rhoen Natur e.V., Friends of the Earth Germany (BUND), Bund Naturschutz in Bayern e.V. (BN), and Federal Agency for Nature Conservation (BfN; Bundesprogramm Biologische Vielfalt – project “Wildkatzensprung”). SM is funded by the DAAD (German Academic Exchange Service), CREOi (Conservation, Research and Education Opportunities International), and the University of California Santa Cruz. AVT received funding from the Karl und Marie Schack-Stiftung. CN received funding from the grant SAW-2011-SGN-3 from the Leibniz Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 23151 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mueller, S.A., Reiners, T.E., Steyer, K. et al. Revealing the origin of wildcat reappearance after presumed long-term absence. Eur J Wildl Res 66, 94 (2020). https://doi.org/10.1007/s10344-020-01433-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-020-01433-7