
Abstract
Noninvasive genetic studies of wild animals enable the recovery of information infeasible to obtain using other means. However, the low quantity and quality of noninvasively collected DNA often challenge the retrieval of reliable genotypes, which may cause biases in downstream analyses. In this study, we optimized SNP (single nucleotide polymorphism) genotyping of fecal samples from moose (Alces alces) with the main purpose of exploring the potential of using noninvasively retrieved genotypes for individual- and sex identification. Fecal pellets were collected during the late winter of 2016 on the Swedish island of Öland in the Baltic Sea and DNA was extracted and genotyped using 86 autosomal, six sex-specific and five species diagnostic SNPs. The SNP error rate of the quality filtered dataset was 0.06 and the probability of identity for siblings below 0.001. Following a thorough quality filtering process, 182 reliable genotypes were obtained, corresponding to 100 unique individuals (37 males, 63 females), with an estimated male proportion of 37% (± 9%). The population size, estimated using two different capture-mark-recapture approaches, was found to be in the range of 115–156 individuals (95% CI). Furthermore, moose on Öland showed significantly lower heterozygosity levels (zHexp = −5.51, N = 69, p Hexp = 3.56·10−8, zHobs = −3.58, N = 69, pHobs = 3.38·10−4) and appeared genetically differentiated from moose on the Swedish mainland. Thus, we show that quality controlled noninvasively derived SNP genotypes can be highly informative for individual and population monitoring in a large ungulate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acquiring reliable data on fundamental parameters such as distribution, population size, and reproduction is important for wildlife research and management alike (Allendorf and Hard 2009; Bender 2006; DeYoung et al. 2006; Morellet et al. 2007). This is particularly true for populations of ecological and economical importance or of conservation concern (Bunnefeld and Keane 2014; Skrbinsek et al. 2012). However, studying wild populations is often logistically demanding, especially for species that range widely, across inaccessible terrain, occur at relatively low densities, and are wary of humans. Many different observational methods have therefore been used to generate such estimates, ranging from spoor and pellet counts to camera-trapping, ringing, collaring, and aerial surveys. These methods may produce good relative population estimates, allowing for comparisons between censuses and monitoring of population trends over time (Balme et al. 2009; Cutler and Swann 1999); however, converting observation frequencies to absolute numbers is typically fraught with biases (Anderson 2001; Anderson 2003; Garel et al. 2010; Lonergan et al. 2013; Ringvall et al. 2000). For example, a lack of individual recognition may lead to counting the same individual repeatedly, thereby inflating the population estimate, a pervasive problem in camera trap based censuses (Treves et al. 2010). In some species individual recognition is feasible, but more commonly the animal has to be fitted with some form of tag, typically requiring capture. An alternative is to use an individual’s genotype; a unique and permanent tag that can be obtained noninvasively, i.e., from remnants such as feces, saliva, or hair (Taberlet and Luikart 1999). Recapture patterns and analyses of individual genotypes allow estimates of a wide range of evolutionary, ecological, and behavioral processes in wild populations (Arandjelovic et al. 2010; Norman and Spong 2015; Palsboll 1999).
Fecal samples are a commonly used source of DNA for noninvasive genetic studies of wildlife (Brinkman et al. 2011; Gray et al. 2014; Hettinga et al. 2012; Kohn et al. 1999; Prugh et al. 2005; Robinson et al. 2009), and their appeal comes from high abundance, ease of identification, and relatively high concentrations of DNA (Morin et al. 2001). Yet, obtaining high quality genetic data from fecal samples collected in field can be challenging (Creel et al. 2003; Fickel and Hohmann 2006; Hebert et al. 2011). In order to obtain precise population estimates from recapture studies, a sufficient number of samples have to be collected, and the sample size is furthermore determined by the outcome of the genotyping process which to a large extent is affected by the quality and quantity of the sampled target DNA. Insufficient amounts of intact target DNA, contamination, and the presence of PCR-inhibitors are recognized problems related with fecal samples, and degradation of DNA over time due to exposure to UV radiation, heat, and humidity, makes the timing of sampling of importance and prompt sampling preferable (Panasci et al. 2011; Rea et al. 2016). Following sampling, the preservation method of the collected material is an additional factor affecting the DNA quality (Panasci et al. 2011; Taberlet and Luikart 1999). Furthermore, for individual-based applications, amplification failure in the form of allelic misprinting can generate false genotypes and/or incorrect matching of samples (Creel et al. 2003), which may cause loss of information due to inconsistent genotypes but also risks biasing downstream applications such as population size estimations and relatedness analyses. PCR-inhibitors, originating from the digestive system (e.g., bile-salts) or from dietary components, have been identified as the main cause of amplification failure for herbivore fecal samples (Ramon-Laca et al. 2015). Among the known PCR-inhibitors in plant material are complex polysaccharides that disturb the enzymatic process and secondary metabolites such as phenolic compounds that bind to DNA (Kim et al. 1997; Schrader et al. 2012). Various approaches have been applied to handle these problems, such as sampling without targeting inhibitors (Hettinga et al. 2012), characterization of target DNA before amplification (Ball et al. 2007), and/or dilution of DNA extracts (Schrader et al. 2012). The quality of the genotyping is furthermore affected by the type of molecular marker used. Here, single nuclear polymorphism (SNP) assays have proven particularly useful for noninvasive studies as they work well on fragmented DNA, which can be explained by the relatively short fragment lengths required for amplification in comparison to for example microsatellite markers (Fabbri et al. 2012; von Thaden et al. 2017). Moreover, liquid-based SNP microarray platforms typically have low copy number detection thresholds which are an important feature when analyzing degraded DNA. As a consequence, SNP markers have been shown to successfully and reliably amplify samples containing low amounts of DNA derived from fecal samples (Kraus et al. 2015; Norman and Spong 2015).
The largest deer species, the moose (Alces alces), plays an important ecological and economical role in boreal regions of the world (Molvar et al. 1993). As a selective browser, the moose feed extensively on leaves and shoots of woody plants including trees that are grown for forestry purposes. Moreover, the moose is an appreciated game species associated with strong cultural values leading to that the moose management has to consider conflicting interests regarding the size and distribution of the population. Sustainable wildlife management requires reliable and current population demographic estimates, especially important for heavily managed populations such as moose in Sweden. Moose are widely distributed throughout the Scandinavian peninsula and about 300,000–350,000 individuals (pre-harvest) are estimated to be found in Sweden, of which about 25–30 % is non-randomly harvested each year (Jägareförbundet 2015). Until now, population demographic parameters are based mainly on hunters’ observations and pellet counts which have been collected yearly for decades and hence provide valuable relative abundance and demographic estimates. Also, aerial inventories are occasionally performed which are efficient for population surveys but very costly. Thus, genetic methods would be a strong complement to traditional population monitoring efforts that, in addition to more accurate population size estimations, could add information about sex-ratio, dispersal, reproduction, and genetic variation. Furthermore, the identification of individuals allows studies of behavioral components, such as resource and space use.
In this study, we evaluate the population monitoring capacity of SNP genotyping of noninvasively collected fecal pellets of moose. During the last decade, the health of moose on the Baltic island of Öland has been investigated. Despite the lack of large predators, lower than normal calf summer survival rates have been confirmed on the island, and it has been found that limited resource availabilities in combination with heat stress are important factors behind the low calf survival (Malmsten 2014). The island is furthermore a suitable study area for retrieving capture-mark-recapture-based demographic estimates, such as population size and sex ratio, since the population can be assumed to be relatively closed, with very few individuals entering or leaving the area during the study period. The last reported aerial inventory, conducted in the beginning of 2005, reported a population size at the time of 150 individuals distributed as 33% males, 47% females, and 20% calves (Jonsson 2007). The hunting pressure on moose on Öland varies between years; from zero harvested animals to a maximum of 43 harvested animals between the years 2000 and 2016, a variation that can be explained by an adaptive management with harvesting levels adjusted according to current evaluations of the population and by voluntary suspended hunting during some years by the owners of the hunting rights. In this study we; (1) evaluate the performance of the moose SNP panel on noninvasively collected fecal samples, and used this data to (2) estimate the population size, sex ratio and (3) explore the genetic differentiation between moose on Öland and the Swedish mainland.
Methods
Sampling and location
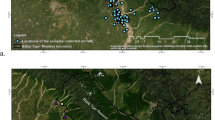
Öland is located in the Baltic Sea, 3–30 km southeast of Sweden (56°N, 16°E). The island covers 1342 km2 (137 km long, 16 km at the widest point). From 29th of January to 11th of April 2016, moose pellets (n samples = 489) were collected following a citizen science approach involving local inhabitants and/or hunters on Öland (Fig 1). Initially, sampling was conducted using a systematic line transect design but the encounter rate proved to be too low. Consequently, the sampling was altered to an opportunistic method meaning that areas preferred by moose (i.e., forest patches where they feed and seek shelter) were scanned for pellets. During the sampling period, moose on Öland were regarded as a demographically and geographically closed population with no births and negligible number of deaths or migrants between the island and the mainland. Similar studies estimating population size from noninvasive sampling have estimated that a necessary sampling effort, taking into account the loss of samples due to genotyping failure, is about three times as many collected samples as there are unique individuals (Kindberg et al. 2011; Puechmaille and Petit 2007). We hypothesized the moose population on Öland to consist of about 150 animals (Jonsson 2007), giving us a target of a minimum of 450 samples. In line with our assumption that the population was closed and since DNA degrades rapidly in field, only fresh fecal pellets were targeted during sampling. For each encountered “pile” of moose fecal pellets, five pellets were collected into a plastic bag, and the samples were stored in freezer until DNA extraction. The sampling effort varied between days (46 days in total) with the number of collected samples ranging between 1–24 samples / day. The climatic conditions during the time of sampling included a mean temperature of 3.4 °C and a mean precipitation per day of 0.6 mm (SMHI 2018). For validation of the sex-specific SNPs, we included seven fecal samples from moose of known sex (two female, five male) collected in enclosures at the zoo “Lycksele djurpark.” Also for validation purposes, high quality DNA extracted from blood samples available from 34 GPS equipped moose on Öland were included in the study. The blood was collected in 2012 and 2015, using EDTA tubes, and stored in −20 °C after collection (Permits from the Committee of Ethical evaluation, Umeå: A116-09, A12-12, A50-12, A205-12). Population genetic differentiation between moose on Öland and its source population; the Swedish mainland, was investigated using genotypes generated in a previous study using the same SNP panel (Blahed et al. 2018).

Map of Sweden with the island of Öland enlarged, showing sampling locations of the fecal pellets included for genotyping
DNA extraction
To target epithelial cells from moose intestine wall present on the surface of the fecal pellets, while minimizing the inclusion of nontarget DNA from dietary compounds, only the surface material of the pellets was used for DNA extraction. From each fecal pellet sample (1 pellet/sample), 0.18–0.22 g surface material was scraped off using a scalpel. The fecal pellets were kept on ice during handling in the lab, and extraction of DNA was conducted manually using the QIAamp DNA Stool Mini Kit and QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the protocols of the manufacturer. The whole blood samples used for validation were thawed on ice in room temperature, and 200 μl of whole blood was used as input volume for automatized extractions using the QIAsymphony SP instrument and the QIAsymphony DSP DNA Mini Kit (Qiagen, Hilden, Germany). DNA extracts were stored at −20 °C.
SNP genotyping and evaluation
Genotyping was performed with SNPs previously developed by reduced representation library high-throughput sequencing of Swedish moose, including two individuals from Öland (Blahed et al. 2018). The SNP panel included 86 autosomal SNPs (minor allele frequency range = 0.25-0.50), six sex-specific SNPs and five species diagnostic mitochondrial SNPs (Online Resource 1). In total, 530 samples were genotyped on the Fluidigm Biomark platform (Fluidigm Corporation, San Fransisco, USA). Based on previous genotyping of high quality DNA (extracted from blood), the estimated error rate for the autosomal SNP panel was below 0.002 per allele and zero for the sex-specific and species diagnostic SNPs, and the panel has also showed to separate first order relatives with high confidence. To minimize the risk of cross-contamination between sources of DNA, our lab features a strict unidirectional flow in which DNA extraction, PCR amplification, and SNP analyses are performed in separate rooms. All rooms have designated equipment and only processed samples are moved between rooms. Every genotyping run included three non-template controls (NTCs), i.e., samples containing water instead of DNA, for detection of primer-dimer binding during the PCR and as a negative contamination control. The genotyping results were evaluated visually using the Biomark Genotyping Analysis Software before further analysis, and the assigning of samples to genotypes was controlled for every SNP locus and ambiguous calls were removed. Also, SNPs that did not produce distinct genotypes were discarded. Our lab has developed and set up SNP panels for more than 10 model systems, and in all, amplification success correlates to error rate (unpublished data). When working with low quality samples, the exact threshold for reliable genotypes is difficult to know in advance, but our lab has found that 75% amplification rate (both alleles called irrespective of their reliability) is an appropriate initial filtering threshold. As a consequence, samples with an amplification rate below 75% (including autosomal SNPs only) were discarded to decrease the number of missing alleles for downstream analyses. Initially, 65 samples were replicated for genotype reliability assessments, and these duplicated samples varied considerably in reliability with the poorest samples producing false unique individuals (ghost individuals) in downstream analyses. To enable assessments of the genotype quality for each sample and to avoid biasing our population size estimate, we attempted to produce duplicates for all samples with an initial amplification rate above 75%. Genotyping errors for the autosomal SNPs were defined as mismatches between assigned alleles of duplicated samples for both heterozygous and homozygous loci (thus potentially including both misprinting and dropouts). The error rate was calculated as the number of allelic mismatches between duplicates divided by the total number of amplified alleles. The unfiltered error rate was estimated based on the total number of genotyped samples with amplification rate above 75%, and the filtered error rate was calculated for the quality filtered genotypes that were included for individual identification. Using the R software (R Core Team 2017), a negative binomial general linear model (GLM) was applied to investigate the relationship between the number of amplified SNPs and the number of mismatches for the duplicated genotypes with an amplification rate above 75%. A likelihood ratio test was performed to test the overall model fit.
Sex identification and species diagnostics
The performance of the sex-specific SNPs on noninvasive fecal samples was examined partly by genotyping fecal samples of known sex, and partly by controlling against matching blood samples from GPS equipped moose of known sex. We tested six sex-specific markers (all on the Y-chromosome), and retained four that gave consistently reliable results. The error rate of the sex-specific SNPs was estimated based on duplicate samples of the quality filtered dataset; duplicated samples identified with different sex were recorded as errors. Using the true dropout rate, calculated from genotyping of known samples, the expected distribution of false results (negatives or positives) versus correct amplification across all loci was established. This allowed us to set amplification result thresholds that produced a sex determination with a confidence exceeding 98%. The binomial 95% confidence interval for the proportion of males detected in the population was computed based on the empirically determined proportion of males and unique individuals. For the species diagnostic SNPs, the error rate was estimated per allele for 65 replicated samples.
Individual identification
The number of unique individuals in the dataset was estimated using the R package allelematch (Galpern et al. 2012). The maximum number of mismatching alleles allowed for separating individuals without matching unique genotypes was investigated with the function amUniqueProfile. In parallel, all sample duplicates were analyzed with the feature amUnique which match genotypes based on dissimilarities between pairs. The outputs of the amUnique analyses with different thresholds for allelic mismatching were screened manually for correct matching of sample duplicates and appearances of false unique individuals (i.e., sample duplicates appearing as two unique individuals). As a result, the duplicated genotypes were initially matched against each other with a maximum allowed mismatch of alleles set at 20 and the genotypes producing false unique individuals were removed to avoid biased population size estimations. Of the 34 individuals for which we had blood samples, 23 were alive at the time of the fecal pellet collection in 2016. The blood genotypes were matched with the quality filtered fecal genotypes in allelematch.
Population size estimation
The population size of moose on Öland was estimated using two different capture-mark-recapture (CMR) approaches. The general assumptions made for the CMR analyses were that the studied population was closed in space and time and that all individuals were correctly genotyped and identified. We obtained a maximum likelihood estimate, with a multinomial probability distribution, using the R package capwire (Pennell et al. 2013). Capwire offers two different models for population size estimation; the first assumes that all individuals are equally likely to have been sampled; the second model takes into account individual-based heterogeneity in the probability of capture. A likelihood ratio test was performed running the lrtCapwire simulation to select the best fitting model for population size estimation. The confidence interval was estimated with the bootstrapCapwire function performing parametric bootstrapping (n = 1000) to obtain 95% confidence intervals for the population estimate. The second CMR approach was a Bayesian single sampling session method based on a non-informative prior distribution (Gazey and Staley 1986; Petit and Valiere 2006). The function CMRpopsize written by Petit and Valiere (2006) was used, which incorporated the following information: total number of genotyped samples, number of uniquely identified individuals, and maximum population size.
Statistics and population genetic differentiation
Basic statistic calculations were performed using the R package BSDA (Evans 2017). Calculations of allele frequencies, Hardy-Weinberg equilibrium (HWE) and probability of identity (PI) were executed in GenAlEx 6.503 (Peakall and Smouse 2006; Peakall and Smouse 2012). These parameters were estimated based on the uniquely identified genotypes of which consensus sequences were retrieved by the amCluster function of allelematch (consensus method = 2) with missing data replaced when available for other identical genotypes. To investigate population genetic differentiation between moose on Öland and the Swedish mainland, expected and observed heterozygosity and F-statistics were calculated in GenAlEx and a covariance-standardized principal coordinate analyses (PCoA) were performed using the same software to visualize similarities or dissimilarities of genotypes.
Results
Evaluation of genotyping
Manual screening of the genotyping output resulted in the exclusion of 12 of the 86 autosomal SNPs in the panel in order to avoid unreliable genotypes as an effect of interpretation difficulties. Three more SNPs with an error rate above 0.20 were excluded, resulting in 71 autosomal SNPs for analyses. For the analyses of population differentiation, additionally two SNPs were removed due to monomorphism and low amplification rate, resulting in 69 autosomal SNPs (Online Resource 1). Calculated for the uniquely identified genotypes, the mean estimated minor allele frequency (MAF) was 0.33, five of the SNPs deviated from HWE (p < 0.05, Online Resource 2). The probability of identity was estimated below 0.001 (p = 7.91 · 10−4) combining eight of the most informative SNPs, and a similar level of probability (p = 7.68 · 10−4) was achieved for first order relatives using 16 SNPs. Excluding replicates, 489 fecal pellet samples were SNP genotyped for autosomal, sex-specific, and species diagnostic markers. Successful amplification results (> 75% of the SNPs) were obtained for 302 of the genotyped samples; hence the rate of successful amplifications was 62%. Out of these, the species diagnostic SNPs revealed eight samples as roe deer. Out of the 34 blood samples included for genotyping, 33 samples amplified successfully. Successful amplification was achieved for 269 fecal sample duplicates that subsequently were included for further evaluations. At this point, the (unfiltered) mean error rate for the autosomal SNPs was estimated to 0.14 (sd = 0.07), varying between 0.03 and 0.35, and the mean sample amplification rate was 0.94 (range 0.76-1.00). The number of amplified SNPs was found to predict the number of mismatching SNPs (Negative binomial GLM: χ2 = 144.89, p < 0.001, Fig 2). Hence, samples with high genotyping success were associated with higher genotyping reliability. An additional quality-filtering step was applied after the observation that genotypes with 30% or higher allelic mismatch produced unreliable genotypes, leading to those 224 samples remained for matching of genotypes (Fig 3). The mean error rate of the quality filtered dataset (used for identification of unique individuals) was 0.06 (sd = 0.04, range 0–0.18) and the mean amplification rate 0.97 (range 0.81–1). See Online Resource 3 for error and amplification rates for single SNPs.

The number of mismatching SNPs and the number of amplified SNPs with a fitted negative binomial GLM curve. The number of mismatching SNPs was predicted by the number of amplified SNPs (p < 0.001). At this stage, 74 SNPs were analyzed and samples with an amplification rate below 75% had been discarded

The main steps of the quality filtering process undertaken for the genotyped samples
Sex-specific and species diagnostic SNPs
The genotyping of fecal samples of known sex gave an estimated drop-out (Y-specific allele not amplifying in male) rate of 0.23 for the four sex-specific SNPs that were constantly informative. No case of allelic drop-in (Y-specific allele amplifying in female) was detected in the validation data; however, amplification of sex-specific SNPs for genotypes determined as female occurred at low levels in the analyzed noninvasive dataset. Based on the empirically found drop-out ratio, the probability of a male genotype amplifying at less than four out of eight sex-specific SNP loci was found less than 0.05 (based on known duplicates; see Fig 4). Thus, considering the low risk of allelic drop-in, genotypes amplified at a minimum of 50% of the sex-specific loci were identified as male, and under these probabilities, the error rate of the sex identification was calculated to below 0.01. The sex identification of the unique individuals was conducted without any contradicting or ambiguous calls, and all cases where GPS collared individuals (blood samples) matched with individuals in the noninvasive dataset confirmed the sex identification performed on the fecal material. The amplification rate for the species diagnostic SNPs included for analysis was estimated to 0.97 and the error rate was 0.02 (sd = 0.04). All errors occurred at the same SNP that for the Öland population was found variable (Online Resource 1). One species diagnostic SNP failed in the genotyping process.

Probability distribution of sex-specific SNP amplification. For a male genotype, the probability of losing information from more than four out of eight sex-specific SNP loci is below 0.05 (calculated for duplicate sample). Drop-in, i.e., incorrect amplification signals from sample without a Y-chromosome are extremely rare and would not exceed the threshold required for calling the sample male
Individual identification
The initial matching of genotypes (n duplicates = 224) resulted in 169 unique individuals, six unclassified genotypes (neither unique nor matches) and 17 multiple matches. Following a manual screening of the output, 42 individuals were discarded due to inconsistent genotype duplicates that would risk an inflated population size estimation. The final dataset is screened and filtered to maximize the reliability of individual genotypes contained 182 single genotypes. At an allowed maximum of 21 allelic mismatches between samples, the quality-filtered dataset had zero unclassified samples and zero multiple matching samples, also no false unique samples were detected. Under these conditions, the number of unique individuals detected was 100 (Fig 3). Notable is that the amUniqueProfile function provided within the allelematch package suggested a maximum of three mismatching alleles leading to 148 unique individuals. However, our manual evaluations based on duplicate genotypes found that such a low threshold inflated the number of false unique individuals by more than four times compared to our designated threshold. Seven out of 23 genotypes from GPS equipped moose were recaptured among the fecal samples.
Estimation of population size and sex ratio
Based on the recapture pattern of the 100 uniquely identified individuals, a likelihood ratio test for model selection resulted in that the null hypothesis (no within population heterogeneity existing in the data) could not be rejected (p = 0.14). Hence, an equal capture model was fitted to the data to obtain a maximum likelihood estimate of the population size resulting in 134 moose ranging between 115–156 individuals within a 95% confidence interval. The Bayesian population size estimation resulted in 135 individuals with a 95% highest posterior density interval (HPD) of 118–156. Thirty-seven of the 100 unique individuals were identified as male leading to a male sex ratio of 37% ± 9% (95% CI) during the late winter of 2016.
Population genetic differentiation
The expected and observed mean heterozygosity for the Öland moose (100 noninvasive genotypes + 33 blood genotypes–7 matching genotypes = 126 Öland genotypes) were 0.43 and 0.41 and for the Swedish mainland moose (n = 57) 0.48 and 0.46 (Hexp and Hobs for single SNPs reported in Online Resource 2). A z-test found significant differences in heterozygosity between moose on Öland and the mainland (zHexp = −5.51, N = 69, p Hexp = 3.56·10-8, zHobs = −3.58, N = 69, pHobs = 3.38·10−4). In a PCoA, a separation between moose on Öland and the mainland appeared that was most pronounced between genotypes from the northern mainland and Öland. Although no clear separation from the rest of the Öland genotypes, an aggregation of moose sampled in the northernmost forest of Öland appeared in the PCoA (Fig 5). The pairwise population Fst between Öland and the mainland was estimated to 0.03. Due to previously found substructuring of moose in Sweden into a northern and a southern subpopulation (Blahed et al. 2018; Wennerstrom et al. 2016), estimations of population differentiation were performed also with the exclusion of moose from the northern mainland. However, the significant difference in heterozygosity between mainland and Öland persisted (zHexp = −2.90, N = 69, p Hexp = 3.79·10−3, zHobs = −2.30, N = 69, pHobs = 0.02), and the Fst between the southern mainland and Öland was again estimated to 0.03 (Table 1 a). The highest Fst, 0.07, was found between moose in the northernmost area of Öland and the northern mainland (Table 1 b). The inbreeding coefficient, F, for the Öland moose was 0.05 and for the southern mainland moose 0.04 (no significant difference).

PCoA visualizing population differentiation between Öland moose (n = 126) and moose from the Swedish mainland (n = 57). The strongest differentiation occurs between Öland moose and the northern mainland subpopulation. PC 1 and 2 explain 7.52% and 5.51% of the variation, respectively
Discussion
We were interested in assessing the performance of our recently developed panel of moose SNP markers on low quality DNA from noninvasively collected fecal samples. Individual identification and sex determination of fecal pellets enable large-scale population genetic assessments at a relatively low economic cost and logistical effort. Following a thorough data screening process, reliable information about species, individual identity, and sex were delivered by the SNP panel. The species diagnostic SNPs showed high amplification success although one SNP surprisingly appeared to be polymorphic among moose on Öland and the inclusion of SNPs for separating deer species proved useful on fecal samples since eight roe deer were detected in the data. Including species diagnostic SNPs will be even more important when genotyping DNA extracted from, e.g., hair or saliva since the species of such samples is difficult or impossible to determine using other methods. The identification of individuals was highly accurate for the quality filtered autosomal SNP genotypes; out of 182 genotypes 100 unique individuals were identified. The estimated population size was consistent between the two different CMR methods used and was in the range of 115–156 individuals. However, the lower than desired sample size (due to lower than expected genotyping success) for the CMR analysis may have decrease the capture probability and consequently affect the precision of the population estimate. Sampling was optimized to include as many individuals as possible from the entire island but after the genotype filtering process, the southernmost area of Öland was not represented. This implies that we failed to cover the whole study area and according to GPS information available (Ericsson et al. 2017; Neumann 2018), at least six moose were stationary on the southernmost part of the island during the winter of 2016. The vegetation pattern on Öland is highly heterogeneous and the vast majority of the preferred moose habitats occur in the north and middle parts of the island, which make the loss of samples from the south less problematic and should not largely affect the population size estimation. Yet, a relatively low “re-capture” rate of GPS collared individuals (seven out of 23) compared to the number of unique individuals (100) and the estimated population size (~ 135) may be suggestive of a too low sample size. However, it may be that collared individuals (with darting from a helicopter) were found in more open areas, whereas sample collection focused on more wooded areas with higher densities of pellets, explaining the low “recapture” rate of collared individuals in the fecal data.
We estimated the male ratio on Öland to 37% (± 9%) at the time of sampling which is within the confidence interval of the hunters’ observation index (Älgdata 2018). For unknown reasons one or two sex-specific SNPs failed to amplify in some of the genotyping runs, hence, including five or six sex-specific SNPs is recommended to ensure a sufficient number for analysis. Nevertheless, the sex identification of the 100 uniquely identified individuals was performed without any contradicting results and can be considered as highly reliable (see Fig 4).
The moose SNP panel has previously shown high amplification success and has produced genotypes with very high reliability when DNA of high quality have been analyzed (Blahed et al. 2018). Among the advantages of the particular SNP genotyping technology used is its sensitivity to low concentrations of target DNA. Based on successful outcomes using fluid-based SNP genotyping of noninvasively collected fecal samples, but for other species (Kraus et al. 2015; Norman and Spong 2015; von Thaden et al. 2017), the expectation was to obtain satisfying results also for DNA extracted from moose fecal samples. Therefore, we were surprised by the low amplification rates and the high variability in genotype quality between samples. However, when dealing with noninvasive samples, there are factors complicating the different stages of the genotyping process. According to Panasci et al. (2011) fecal samples should be collected within one week or less after defecation for acceptable amplification result. Although fresh samples were targeted in this study, the exact age of the fecal pellets could not be determined; hence the DNA might have been largely degraded in some samples. Moreover, we suspect that the presence of PCR-inhibitors such as phenolic compounds, in particular pronounced in the winter diet of moose, contributed to the low amplification rates and affected the genotyping quality resulting in low genotype reliability. The initial threshold for satisfying amplification was set at 75% SNP amplification, which in our lab, has been shown as valid for the error rate and PI of other species (unpublished data). The final mean sample amplification rate for the processed dataset was 97% showing that a higher threshold is required for moose winter fecal data. The relationship between sample amplification rate and genotypic reliability, shown by the GLM, suggests that a high amplification threshold would prevent the need for analyzing sample replicates for assessment of genotype quality. This threshold appears to vary between species, diets, fecal properties and/or local environment. To increase the target-to-total DNA ratio and to decrease the PCR-inhibitor concentrations, Ramon-Laca et al. (2015) found that for herbivorous species, swabbing the fecal pellets in the field and extract DNA from buffer solution gave better results compared to sampling and extraction methods similar to ours. Collection of whole fecal pellets is a straightforward sampling method suitable for citizen-science-based approaches but in order to optimize the recovery of reliable genotypes for analysis, another sampling strategy might be preferable. Notable is that the diet of moose changes between seasons, and it is possible that the genotyping success would be affected if sampling would be conducted during a different time of the year.
The filtering process undertaken in this study aimed for reliable genotypes that would result in a conservative population assessment, consequently, our best estimate was 134–135 moose ranging between 115–156 individuals. We find this approach appropriate both in terms of method development; in order to understand and optimize the process of analysis, and also from an animal ethical perspective since we are studying a species that is quite heavily controlled by hunting harvest. The flight inventory made in 2005 detected 150 individuals (Jonsson 2007), which lies within the confidence interval of our estimation. Obviously, a more recent population count would have been preferable for a comparison with our method. In addition to the demographic information obtained, and despite the high MAF of the SNP panel, genetic differentiation that goes further than the previously found north–south separation of Swedish moose (Blahed et al. 2018; Charlier et al. 2008; Wennerstrom et al. 2016) was detected. We found genetic differentiation between moose on Öland and moose on the mainland including significantly lower heterozygosity levels among Öland moose. Hence, although movement of moose between the mainland and Öland is possible and occasionally occur, since moose are able to swim between the mainland and the island, the gene flow is probably limited. Moreover, the lower heterozygosity levels among Öland moose could be an indication of a founder effect, which also is supported by the fact that no indication of inbreeding was detected. The tendency of higher similarity of moose in the northernmost forest area of Öland compared to moose in remaining areas of the island is somewhat surprising due to the limited size of the island. However, the northernmost forest is the largest continuous forest habitat on Öland, and moose in this area may have their resource demand covered within this forest, whereas moose in more fragmented parts of the island need to move and reproduce over larger distances, possibly creating a weak genetic differentiation, at least during winter. In conclusion, we show that reliable and highly informative genotypes can be obtained from noninvasive moose fecal samples. Thus, SNP genotyping is now an available tool for noninvasive population monitoring of moose, including population demography, large-scale population connectivity and structure, as well as pedigree-based work to study individual dispersal and reproductive success.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.
References
Älgdata (2018) https://algdata-apps.lansstyrelsen.se/algdata-apps-stat. Accessed 2018-03-20
Allendorf FW, Hard JJ (2009) Human-induced evolution caused by unnatural selection through harvest of wild animals. Proc Natl Acad Sci U S A 106:9987–9994. https://doi.org/10.1073/pnas.0901069106
Anderson DR (2001) The need to get the basics right in wildlife field studies. Wildl Soc Bull (1973-2006) 29:1294–1297
Anderson DR (2003) Response to Engeman: index values rarely constitute reliable information. Wildl Soc Bull (1973-2006) 31:288–291
Arandjelovic M, Head J, Kuhl H, Boesch C, Robbins MM, Maisels F, Vigilant L (2010) Effective non-invasive genetic monitoring of multiple wild western gorilla groups. Biol Conserv 143:1780–1791. https://doi.org/10.1016/j.biocon.2010.04.030
Ball MC et al (2007) Characterization of target nuclear DNA from faeces reduces technical issues associated with the assumptions of low-quality and quantity template. Conserv Genet 8:577–586. https://doi.org/10.1007/s10592-006-9193-y
Balme GA, Hunter LTB, Slotow R (2009) Evaluating methods for counting cryptic carnivores. J Wildl Manag 73:433–441. https://doi.org/10.2193/2007-368
Bender LC (2006) Uses of herd composition and age ratios in ungulate management. Wildl Soc Bull 34:1225–1230. https://doi.org/10.2193/0091-7648(2006)34[1225:uohcaa]2.0.co;2
Blahed IM, Konigsson H, Ericsson G, Spong G (2018) Discovery of SNPs for individual identification by reduced representation sequencing of moose (Alces alces). Plos One:13. https://doi.org/10.1371/journal.pone.0197364
Brinkman TJ, Person DK, Chapin FS, Smith W, Hundertmark KJ (2011) Estimating abundance of Sitka black-tailed deer using DNA from fecal pellets. J Wildl Manag 75:232–242. https://doi.org/10.1002/jwmg.22
Bunnefeld N, Keane A (2014) Managing wildlife for ecological, socioeconomic, and evolutionary sustainability. Proceedings of the National Academy of Sciences of the United States of America 111:12964–12965. https://doi.org/10.1073/pnas.1413571111
Charlier J, Laikre L, Ryman N (2008) Genetic structure and evidence of a local bottleneck in moose in Sweden. J Wildl Manag 72:411–415. https://doi.org/10.2193/2007-122
Creel S et al (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009. https://doi.org/10.1046/j.1365-294X.2003.01868.x
Cutler TL, Swann DE (1999) Using remote photography in wildlife ecology: a review. Wildl Soc Bull 27:571–581
DeYoung RW, Demarais S, Honeycutt RL, Gee KL, Gonzales RA (2006) Social dominance and male breeding success in captive white-tailed deer. Wildl Soc Bull 34:131–136. https://doi.org/10.2193/0091-7648(2006)34[131:sdambs]2.0.co;2
Ericsson G, Malmsten, J., Neumann, W., Dettki, H., Nilsson, K., Stenbacka, F., Evans, A., Arnemo, J., Edenius, L., Cromsigt, J., Singh, N. (2017) Årsrapport GPS-älgarna Öland 2016/2017; Rörelse, reproduktion. Institutionen för Vilt, Fisk och Miljö. Sveriges Lantbruksuniversitet., Umeå
Evans ATAaB(2017) BSDA: Basic Statistics and Data Analysis., 1.2.0 edn
Fabbri E et al (2012) Comparison of single nucleotide polymorphisms and microsatellites in non-invasive genetic monitoring of a wolf population. Arch Biol Sci 64:321–335. https://doi.org/10.2298/abs1201321f
Fickel J, Hohmann U (2006) A methodological approach for non-invasive sampling for population size estimates in wild boars (Sus scrofa). Eur J Wildl Res 52:28–33. https://doi.org/10.1007/s10344-005-0003-5
Galpern P, Manseau M, Hettinga P, Smith K, Wilson P (2012) Allelematch: an R package for identifying unique multilocus genotypes where genotyping error and missing data may be present. Mol Ecol Resour 12:771–778. https://doi.org/10.1111/j.1755-0998.2012.03137.x
Garel M, Bonenfant C, Hamann JL, Klein F, Gaillard JM (2010) Are abundance indices derived from spotlight counts reliable to monitor red deer Cervus elaphus populations? Wildl Biol 16:77–84. https://doi.org/10.2981/09-022
Gazey WJ, Staley MJ (1986) Population estimation from mark-recapture experiments using a sequential bayes algorithm. Ecology 67:941–951. https://doi.org/10.2307/1939816
Gray TNE, Vidya TNC, Potdar S, Bharti DK, Sovanna P (2014) Population size estimation of an Asian elephant population in eastern Cambodia through non-invasive mark-recapture sampling. Conserv Genet 15:803–810. https://doi.org/10.1007/s10592-014-0579-y
Hebert L, Darden SK, Pedersen BV, Dabelsteen T (2011) Increased DNA amplification success of non-invasive genetic samples by successful removal of inhibitors from faecal samples collected in the field. Conserv Genet Resour 3:41–43. https://doi.org/10.1007/s12686-010-9280-8
Hettinga PN, Arnason AN, Manseau M, Cross D, Whaley K, Wilson PJ (2012) Estimating size and trend of the North Interlake woodland caribou population using fecal-DNA and capture-recapture models. J Wildl Manag 76:1153–1164. https://doi.org/10.1002/jwmg.380
Jägareförbundet S (2015) Vilt & Vetande. Svenska jägareförbundet. Accessed 2019-05-12 2019
Jonsson F (2007) Moose management on Öland. A Review of the Management Plans and the Hunting Strategy., Swedish University of Agricultural Sciences
Kim CS, Lee CH, Shin JS, Chung YS, Hyung NI (1997) A simple and rapid method for isolation of high quality genomic DNA from fruit trees and conifers using PVP. Nucleic Acids Res 25:1085–1086. https://doi.org/10.1093/nar/25.5.1085
Kindberg J, Swenson JE, Ericsson G, Bellemain E, Miquel C, Taberlet P (2011) Estimating population size and trends of the Swedish brown bear Ursus arctos population. Wildl Biol 17:114–123. https://doi.org/10.2981/10-100
Kohn MH, York EC, Kamradt DA, Haugt G, Sauvajot RM, Wayne RK (1999) Estimating population size by genotyping faeces. Proc R Soc B Biol Sci 266:657–663. https://doi.org/10.1098/rspb.1999.0686
Kraus RHS et al (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour 15:295–305. https://doi.org/10.1111/1755-0998.12307
Lonergan M, Duck C, Moss S, Morris C, Thompson D (2013) Rescaling of aerial survey data with information from small numbers of telemetry tags to estimate the size of a declining harbour seal population. Aquat Conserv Mar Freshw Ecosyst 23:135–144. https://doi.org/10.1002/aqc.2277
Malmsten J (2014) Reproduction and health of moose in southern Sweden. Swedish University of Agricultural Sciences
Molvar EM, Bowyer RT, Vanballenberghe V (1993) Moose herbivory, browse quality, and nutrient cycling in an Alaskan treeline community. Oecologia 94:472–479. https://doi.org/10.1007/bf00566961
Morellet N et al (2007) Indicators of ecological change: new tools for managing populations of large herbivores. J Appl Ecol 44:634–643. https://doi.org/10.1111/j.1365-2664.2007.01307.x
Morin PA, Chambers KE, Boesch C, Vigilant L (2001) Quantitative polymerase chain reaction analysis of DNA from noninvasive samples for accurate microsatellite genotyping of wild chimpanzees (Pan troglodytes verus). Mol Ecol 10:1835–1844. https://doi.org/10.1046/j.0962-1083.2001.01308.x
Neumann W (2018)
Norman AJ, Spong G (2015) Single nucleotide polymorphism-based dispersal estimates using noninvasive sampling. Ecol Evol 5:3056–3065. https://doi.org/10.1002/ece3.1588
Palsboll PJ (1999) Genetic tagging: contemporary molecular ecology. Biol J Linn Soc 68:3–22. https://doi.org/10.1111/j.1095-8312.1999.tb01155.x
Panasci M, Ballard WB, Breck S, Rodriguez D, Densmore LD, Wester DB, Baker RJ (2011) Evaluation of fecal DNA preservation techniques and effects of sample age and diet on genotyping success. J Wildl Manag 75:1616–1624, 1619. https://doi.org/10.1002/jwmg.221
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pennell MW, Stansbury CR, Waits LP, Miller CR (2013) Capwire: a R package for estimating population census size from non-invasive genetic sampling. Mol Ecol Resour 13:154–157. https://doi.org/10.1111/1755-0998.12019
Petit E, Valiere N (2006) Estimating population size with noninvasive capture-mark-recapture data. Conserv Biol 20:1062–1073. https://doi.org/10.1111/j.1523-1739.2006.00417.x
Prugh LR, Ritland CE, Arthur SM, Krebs CJ (2005) Monitoring coyote population dynamics by genotyping faeces. Mol Ecol 14:1585–1596. https://doi.org/10.1111/j.1365-294X.2005.02533.x
Puechmaille SJ, Petit EJ (2007) Empirical evaluation of non-invasive capture-mark-recapture estimation of population size based on a single sampling session. J Appl Ecol 44:843–852. https://doi.org/10.1111/j.1365-2664.2007.01321.x
R Core Team RFfSC (2017) R: a language and environment for statistical computing
Ramon-Laca A, Soriano L, Gleeson D, Godoy JA (2015) A simple and effective method for obtaining mammal DNA from faeces. Wildl Biol 21:195–203. https://doi.org/10.2981/wlb.00096
Rea RV, Johnson CJ, Murray BW, Hodder DP, Crowley SM (2016) Timing moose pellet collections to increase genotyping success of fecal DNA. J Fish Wildl Manag 7:461–466. https://doi.org/10.3996/112015-jfwm-115
Ringvall A, Patil GP, Taillie C (2000) A field test of surveyors’ influence on estimates in line transect sampling. For Ecol Manag 137:103–111. https://doi.org/10.1016/S0378-1127(99)00319-9
Robinson SJ, Waits LP, Martin ID (2009) Estimating abundance of American black bears using DNA-based capture-mark-recapture models. Ursus 20:1–11. https://doi.org/10.2192/08gr022r.1
Schrader C, Schielke A, Ellerbroek L, Johne R (2012) PCR inhibitors - occurrence, properties and removal. J Appl Microbiol 113:1014–1026. https://doi.org/10.1111/j.1365-2672.2012.05384.x
Skrbinsek T, Jelencic M, Waits L, Kos I, Jerina K, Trontelj P (2012) Monitoring the effective population size of a brown bear (Ursus arctos) population using new single-sample approaches. Mol Ecol 21:862–875. https://doi.org/10.1111/j.1365-294X.2011.05423.x
SMHI SMaHI (2018) Meteorological observations data downloads. https://opendata-download-metobs.smhi.se/explore/. Accessed 2018-04-10
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Linn Soc 68:41–55. https://doi.org/10.1111/j.1095-8312.1999.tb01157.x
Treves A, Mwima P, Plumptre AJ, Isoke S (2010) Camera-trapping forest–woodland wildlife of western Uganda reveals how gregariousness biases estimates of relative abundance and distribution. Biol Conserv 143:521–528. https://doi.org/10.1016/j.biocon.2009.11.025
von Thaden A, Cocchiararo B, Jarausch A, Jüngling H, Karamanlidis AA, Tiesmeyer A, Nowak C, Muñoz-Fuentes V (2017) Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci Rep:7. https://doi.org/10.1038/s41598-017-10647-w
Wennerstrom L, Ryman N, Tison J-L, Hasslow A, Dalen L, Laikre L (2016) Genetic landscape with sharp discontinuities shaped by complex demographic history in moose (Alces alces). J Mammal 97:1–13. https://doi.org/10.1093/jmammal/gyv146
Acknowledgments
We greatly acknowledge all local actors and our field personnel, especially Kent Nilsson. For assistance in the laboratory we thank Helena Königsson. We also thank Scott Creel and two anonymous reviewers for helpful comments on the manuscript.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This study was funded by the Swedish university of agricultural sciences (SLU), the Swedish National Veterinary Institute (SVA), two local chapters of the Swedish Association for Hunting and Wildlife Management (SAHWM) and the Lennander foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Blåhed, IM., Ericsson, G. & Spong, G. Noninvasive population assessment of moose (Alces alces) by SNP genotyping of fecal pellets. Eur J Wildl Res 65, 96 (2019). https://doi.org/10.1007/s10344-019-1337-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-019-1337-8