
Abstract
Since the nineteenth century, Douglas-fir seed sources have been widely used for establishment of forest stands outside its natural distribution range. In Europe, some of these old Douglas-fir stands are registered as seed stands and provide seed sources for nurseries, although it is unclear from which region in North America they originate. In recent years, the interest in planting Douglas-fir has increased substantially because the species is seen as a potential adaptation option to climate change. This makes the assignment of European Douglas-fir stands of unknown seed origin to their geographic origin in North America increasingly important, because the genetic quality of these plantations must be guaranteed. In this study, we use 13 nuSSR loci to investigate the origin of 67 Austrian and German Douglas-fir stands of unknown origin. We performed a hierarchical Bayesian cluster analysis using 38 native Douglas-fir populations. The resulting clusters are used as reference populations to assign the 67 Central European Douglas-fir stands from Austria and Germany planted more than 80 years ago. Our results suggest that the majority of our investigated Douglas-fir stands come from central Washington (USA), the recommended seed zones for Central Europe. Some stands were located outside the suggested area, e.g. central Oregon and Santa Fe (New Mexico). The accuracy assessment of our approach revealed the best performance for the highest hierarchical level, e.g. assigning populations either to the coastal or the Rocky Mountain variety. As expected, the uncertainty increases with decreasing hierarchical level. The final assessment, if an admixture of seed sources within the European Douglas-fir stands is evident suggests that 23 of the Douglas-fir stands show an admixture which was not detected in our Douglas-fir reference populations growing in the natural distribution range.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Europe has introduced Douglas-fir 190 years ago. In the first 140 years, the importance of the geographic origin of seeds or provenance was neglected and thus, Douglas-fir plantations established prior to the 1980s are mostly of unknown origin (Bastien et al. 2013). However, a number of these “unknown” populations were selected and registered as seed stands mainly because of their superior growth performance. Today, these “unknown” stands as well as “established” plantations are the main European Douglas-fir seed sources (e.g. Kohl and Nather 1991; Konnert 2009; Rau 2009).
The use of the best provenance is important for achieving high productivity rates and to ensure a high adaptive capacity. Provenance trials have shown that not only height and diameter growth increment, but also adaptive relevant characters are under strong genetic control. Adaptive characters include, for example, water-use efficiency effecting the drought tolerance (Darychuk et al. 2012) and bud phenology, which is related to frost hardiness (Eckert et al. 2009). In addition, a shift in the suitable plantation regions within Europe by provenance is expected due to climate change (Chakraborty et al. 2016). Consequently, the origin of European Douglas-fir stands, and particularly those that serve as seed stands, are of high interest.
Douglas-fir [Pseudotsuga menziesii (Mirb.) Franco] is a North-Western American coniferous tree species covering a large latitudinal (19° to 55°) and elevational range (from sea level to 3260 m) (Hermann and Lavender 1990; Kleinschmit and Bastien 1992). Douglas-fir was introduced to many countries worldwide (e.g. New Zealand, Chile, Argentina, Australia, France), mainly because of its growth potential, robustness and timber quality (Holubčík 1968). In Europe, Douglas-fir covers about 750,000 ha, which is the second largest forest area of a non-native conifer tree species (Köble and Seufert 2001) and the largest Douglas-fir area outside its native range (Bastien et al. 2013). In recent years, Douglas-fir has become increasingly interesting because of its growth rates and its resilience versus drought stress. Douglas-fir is considered an additional potential species for adapting European forests to expected changes in climate (IPCC 2007; Eilmann et al. 2013).
The two varieties of Douglas-fir, the coastal (P. menziesii var. menziesii) and the Rocky Mountain (P. menziesii var. glauca) variety, can be distinguished based on their morphological (Allen 1960; Dunlap 1964), population genetic (Li and Adams 1989; Klumpp 1999; van Loo et al. 2015; Neophytou et al. 2016), physiological (Pharis and Ferrell 1966; Lavender and Overton 1972) and growth differences (Kleinschmit and Bastien 1992; Chakraborty et al. 2015). Genetic and paleobotanical evidence supports that the two varieties diverged due to geographic isolation during Pleistocene glacial periods (Gugger and Sugita 2010; Gugger et al. 2010; van Loo et al. 2015). The post-glacial colonisation from their refuges resulted in inter-varietal contact and admixture zones located east of the Cascades of Oregon and Washington (Eckert et al. 2009; Gugger et al. 2010; Wei et al. 2011; van Loo et al. 2015) including a 450 km hybrid zone in British Columbia (Gugger et al. 2010; van Loo et al. 2015).
Within its natural distribution area, Douglas-fir grows on aFalush wide range of site conditions. This has led to provenance trials in several countries across Europe (Barner 1973; Rehfeldt 1979, 1983; Stephan 1980; Campbell 1991; Kleinschmit and Bastien 1992; Konnert and Ruetz 2006; Weißenbacher 2008; Eilmann et al. 2013), aiming to explore the growth properties of different provenances under various climatic conditions. These studies showed differences among the two varieties and within the intravarietal level. In Central Europe, the coastal variety and provenances from Oregon to the west of the Cascade Range show the best growth performance. The Rocky Mountain variety is only recommended for some European countries with continental climate influence, e.g. in countries like Sweden, Finland or Slovakia (Holubčík 1974, 1980; Martinsson 1990; Tigerstedt 1990).
With representative population data from the original distribution range of a species (Cornuet et al. 1999), it is possible to assign populations or individuals of unknown origin to their region of origin (Cornuet et al. 1999) using highly variable molecular markers such as microsatellites (Simple Sequence Repeats; SSRs). Such assignment methods have often been used for tracing back illegal timber trade activities (Lowe and Cross 2011; Jolivet and Degen 2012; Degen et al. 2013; Nazareno and Dos Reis 2014). The accuracy of such methods depends on the genetic differentiation among the reference populations (Cornuet et al. 1999). Thus, it is important that any investigation starts with a genetic clustering and differentiation for the reference population prior to the assignment test. Pooling populations to one cluster based on unsupervised clustering methods may improve the accuracy of the method (Baudouin et al. 2004; García et al. 2006; Nazareno and Dos Reis 2014).
Population genetic analyses of European Douglas-fir stands have differentiated between the two varieties (Konnert and Ruetz 2006; Fussi et al. 2013) but have never used reference populations from its native range (Konnert and Ruetz 2006; Fussi et al. 2013). Given the large ecological and growth differences of provenances within a variety, a refinement in the assignment to intravarietal origins is of interest.
The purpose of this study is to take advantage of a large set of genotyped reference Douglas-fir populations from the native range and develop a new assignment method based on a marker set of 13 nuSSRs to trace back the native geographic origin of 67 Austrian and German Douglas-fir stands of unknown origin, mainly planted more than 100 years ago. Our reference dataset consists of 38 Douglas-fir populations covering the entire distribution range of the coastal and Rocky Mountain varieties across USA and Canada. We are specifically interested in
-
(i)
the pattern of genetic differentiation (clustering) among reference populations within the native range of Douglas-fir,
-
(ii)
the accuracy of the assignment in relation to the inferred clusters at different hierarchical levels, and
-
(iii)
the assignment of 67 Austrian and German Douglas-fir seed stands of unknown origin including the identification of potentially admixed (i.e. seed source mixed) populations.
Materials and methods
For our analysis, two different datasets are used: (i) reference data covering the full range of the natural distribution of Douglas-fir, and (ii) Douglas-fir stands from Austria and Germany of unknown origin. Note for each of these data, the same DNA data extraction method and genotyping was applied.
Study populations
Reference data from USA and Canada
We analyzed 766 individuals from 38 populations (R01-R30, R32-R39, note R31 is missing), covering the natural distribution range of Douglas-fir within the US and Canada (Table 1; Fig. 1; van Loo et al. 2015). A higher sampling density was chosen for Oregon (OR) and Washington (WA) because these areas were traditionally the main seed collection zones for Douglas-fir plantations in Europe (Breidenstein et al. 1990). For each reference population (except population R07 with 18 individuals), 20–22 individual trees were randomly sampled. For 21 of our 38 populations, cambium samples from provenance trials were provided by the Austrian Research Centre for Forests (BFW), Vienna, Austria. These trials used either seeds from the IUFRO-seed collection section (Barner 1973), or seeds collected in natural populations in the US (see Chakraborty et al. 2015). The seed for the remaining 17 populations, were obtained directly from the USDA Forest Service-Placerville Nursery in the US, and the British Columbia Forest Service, Canada. These seeds were cultivated for 2–3 months in the nursery until the seedlings reached approximately 5 cm in height.

Distribution map of Douglas-fir populations (R01–R39) within its natural range in Northwest America
Populations of unknown origin
Populations of unknown origin comprise 67 adult Douglas-fir stands in Austria and Germany (Table 1). In the following, we will refer to these stands as European stands. The oldest of these stands were established in the nineteenth century. Again, cambium samples of 10–26 adult trees per stand were randomly collected, so in sum we obtained samples of 1469 different individuals. Whenever possible, trees with high phenotypic variation were selected to ensure that both Douglas-fir varieties are captured in case adult stands originate from mixed seed collections.
DNA extraction and genotyping
Collected material (cambium samples and cultivated seedlings) of our 38 Douglas-fir reference populations (R1–R30 and R32–R39) and the 67 European Douglas-fir stands was dried and stored in silica gel prior to DNA extraction. The DNA was extracted from leaf or cambium tissue using an OMEGA E.Z.N.A Plant DNA Kit according to the manufacturer’s instructions. For the population assignment, 17 dinucleotide nuSSRs from Slavov et al. (2004) were tested on a set of 16 trees to establish a reliable set of nuclear SSRs with sufficiently high polymorphism.
These tests resulted in 13 highly polymorphic and unlinked nuSSRs (Slavov et al. 2004) which were arranged into four multiplex PCR combinations (Online Resource 1). For further details on SSR selection, we refer to van Loo et al. (2015). Polymerase chain reaction (PCR) amplifications were performed with the QIAGEN Type-it Microsatellite PCR Kit following the manufacturer’s protocol and annealing temperature resulting from the testing (Online Resource 1). PCRs resulting in missing values were repeated using a touch-down procedure with annealing temperatures of 55–45 °C for 10 cycles, each 90 s, followed by 30 cycles at 50 °C, each 60 s. NuSSRs genotypes were resolved on an ABI PRISM™ 3100 DNA Genetic Analyzer (PE Applied Biosystems. Inc.). The sizing of fragments was carried out with a Genescan 3.7 and Genotyper® 2.0 software (PE Applied Biosystems.Inc.), utilising the internal GENESCAN™-500 ROX™ Size Standard (PE Applied Biosystems. Inc.). Allele binning was carried out automatically using the software TANDEM (Matschiner and Salzburger 2009) for both American reference and European populations.
The reference populations are identical to those used in the study by van Loo et al. (2015), where the allele binning was performed manually. Here, we repeat allele binning using an automated method (TANDEM) to optimize the consistency of scoring. The resulting genotypes after allele binning were used for data analysis.
Data analysis and results
Population structure within the reference populations
We started our analysis by performing a Bayesian cluster analysis of the 38 reference populations from North America using the software STRUCTURE v.2.3.4 (Pritchard et al. 2000; Falush et al. 2003, 2007) to infer genetic differentiation and genetic relationships among them. The applied software STRUCTURE uses Markov Chain Monte Carlo procedures (MCMC) for assigning individuals to each one of K predefined clusters based on the individuals’ multilocus genotype. The program assigns each individual a membership proportion (Q value) to each one of the clusters. For all runs, we chose the admixture model and correlated allele frequencies. We performed 20 independent runs for each K between 1 and 10. For each run, we applied 50,000 burn-in replications followed by 100,000 MCMC iterations. We repeated the procedure using the locprior option (Hubisz et al. 2009) because this option is considered to be sensitive in detecting minor differences in the genetic structures which we expected for populations within the coastal variety (Li and Adams 1989; Hubisz et al. 2009; Krutovsky et al. 2009). The STRUCTURE analysis runs were performed with the program STRAUTO v1.0, which allows automation and parallelization of multiple STRUCTURE runs (Chhatre and Emerson 2017).
We defined the optimal number of clusters K, based on two criteria: (i) Maximisation of the statistic ΔK (Evanno et al. 2005), which is based on the second order rate of change of log likelihood of the data for consecutive K values. According to the Evanno et al. (2005), the K value corresponding to the maximum ΔΚ denotes the uppermost hierarchical level. For ΔΚ calculation we used the program STRUCTURE HARVESTER (Earl and von Holdt 2012). (ii) Unimodality among runs for a particular K, i.e. each one of the 20 runs for a particular K should lead to the same clustering solution. We controlled this by processing the multiple runs using the online platform CLUMPAK (Kopelman et al. 2015).
Performing a single cluster analysis for the whole sample may underestimate the “true” number of clusters, because smaller clusters may remain undetected, especially if sampling intensity varies among clusters and / or if hierarchical phylogenetic structure occurs within clusters (Neophytou 2014; Janes et al. 2017; Wang 2017). We performed the hierarchical analysis by repeating the Bayesian clustering within the inferred clusters, as recommended by Pritchard et al. (2010) and Vähä et al. (2007). Finally, we addressed label switching among clusters and average Q values among runs, by applying CLUMPAK after each analysis. CLUMPAK implements the default algorithms of the CLUMPP method (Jakobsson and Rosenberg 2007).
We performed the hierarchical analysis according to the following steps: (i) chose the optimal K value as described above, (ii) assign populations to each one of the K-clusters, using a threshold value of Q > 0.60, (iii) check whether the clustering pattern with the use of locprior was consistent with the analysis without locprior; (iv) If the analysis with locprior option led to the same clustering pattern as without any prior, we reiterated the procedure within each of the resulting K-clusters.
We stopped the procedure (a) if each single population was assigned to one single cluster (i.e. no longer possible to infer further population genetic structure), (b) if populations were not split into at least two clusters, (c) if there was inconsistency of the clustering pattern with locprior versus without prior (which indicates that population genetic structure is very weak).
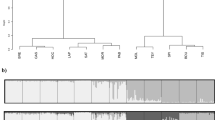
The implementation of the hierarchical cluster analysis as described is shown in Fig. 2, which was created with the software DISTRUCT (Rosenberg 2004). The first analysis including all samples resulted in the separation of the two varieties. We defined this hierarchical level of clustering as ‘hierarchical level 1’ (HL1). Subsequently, the varieties were subdivided into two further clusters, each at the ‘hierarchical level 2’ (HL2; clusters 1–4). After finishing the iterative procedure, we were left with twelve clusters (I–XII) across both varieties at the ‘hierarchical level 3’ (HL3). The spatial distribution of clusters in the native range for each hierarchical level is presented in Fig. 3.

Results of the hierarchical Bayesian cluster analysis of North American populations. Within bar plots, each derived subpopulation (cluster) is marked with a different color. Each population forms a box delimited by horizontal black lines (population designations R01–39). Each individual is represented with a horizontal bar. To the right of each bar plot, the resulting clusters at the corresponding hierarchical level (HL) are lettered (HL1: coastal variety and Rocky Mountain variety, HL2: 1–4, HL3: I–XII). Coastal Douglas-fir populations were sorted from south to north, Rocky Mountain populations from north to south. On the right-hand side, the state is marked where each population is located. CA California, OR Oregon, WA Washington, BC British Columbia, ID Idaho, MT Montana, CO Colorado, NM New Mexico, AZ Arizona

Distribution map of the four hierarchical levels. Map HL1 shows the location of the coastal (red area) and Rocky Mountain (blue area) variety populations of the hierarchical level one. The four clusters with their corresponding populations of hierarchical level two are shown in map HL2. HL3 contains the 12 genetic clusters of hierarchical level three. HL4 shows the populations within the clusters of HL3 and are equitable to hierarchical level four. Three reference populations (R16, R18, R37) are cluster-admixed in this level
Accuracy assessment through self-assignment tests within the inferred clusters
An important step of our analysis is to assess the accuracy of the assignment to clusters/populations at each hierarchical level. Clusters at a higher hierarchical level display a higher genetic differentiation which suggests a higher accuracy in the assignment. For example, a European Douglas-fir stand can be assigned more accurately to one of the two varieties (coastal or Rocky Mountain) as well as to one of the four major clusters (HL2), than to a single reference population within a variety.
We implemented the self-assignment tests for genotypes of known origin (North American) prior to the assignment of the unknown individuals/populations to assess the assignment accuracy for each of the aforementioned hierarchical levels. The software GENECLASS2 (Piry et al. 2004) was applied and each individual tree was assigned to one of the reference populations. Individuals to be assigned are excluded from the reference dataset (leave-one-out-approach) during the computation. This resulted in a percentage of correctly assigned individuals (CA) and a Quality Index (QI) defined as the mean value of the assignment scores of each individual within the population from which it was sampled (Piry et al. 2004).
We ran the self-assignment tests by computing the probability of an individual tree to belong to a population within the hierarchical levels using the computation criteria according to Rannala and Mountain (1997). The simulation algorithm of Paetkau et al. (2004) with a resampling of 10,000 simulated individuals and a Type I error (alpha) of 0.01 (Cornuet et al. 1999; Paetkau et al. 2004) was used.
We repeated the analyses at all hierarchical levels. At HL1, we defined two reference populations corresponding to each one of the two varieties based on the results of the STRUCTURE analysis (see Fig. 2). At HL2, we performed the tests within each variety. For the Rocky Mountain variety, we defined clusters 3 and 4 as reference populations. For the coastal varieties, we defined 3 reference populations: clusters 1 and 2, as well as population R37, which was intermediate between these two clusters. At HL3, we carried out the tests within each one of the clusters 1–4. Again, we treated intermediate populations as additional reference populations. For instance, within cluster 1, there were three reference populations: ‘I’, ‘II’ and R16, which was intermediate between ‘I’ and ‘II’ (see Fig. 2). In addition, we defined an additional hierarchical level (HL4) where single populations within each one of the 12 clusters I-XII from HL3 were considered as separate reference samples. Finally, we performed the analysis with all 38 populations from our North American reference dataset, considering no hierarchical subdivision.
For all GENECLASS2 computations, the total number of 13 nuSSRs needed to be reduced for the reference populations within the southern Rocky Mountain clusters XI and XII. Here, only 10 loci with reliable genotypes were available, because the loci 5A8, 3F1 and 2D6 revealed a high number of missing values. These loci contribute strongly to the differentiation to all other population clusters. The results of the self-assignment analysis are presented in Fig. 4.

Arrangement of the four hierarchical levels (HL1–HL4) starting from 38 populations of the reference data set grouped into the coastal and Rocky Mountain variety, followed by four and twelve genetic clusters. For each branch of the top-down procedure, the Quality index (QI) and the correctly assigned individuals (CA) were calculated with the self-assignment test in GENECLASS2. Populations marked with an X between two clusters are indicated as cluster admixed populations
Origin assignment of European Douglas-fir
The final step of our study was to “trace back” and identify the seed origin of the 67 European Douglas-fir stands listed in Table 1. We performed “assignment of groups of individuals” with the software GENECLASS2 (Piry et al. 2004). We applied the multilocus Bayesian criterion according to Rannala and Mountain (1997) with a threshold value of p = 0.05 for all runs. We carried out separate analysis at each hierarchical level. The reference populations for each analysis run followed the hierarchical order of the self-assignment test as shown in Fig. 3.
Prior to the assignment with GENECLASS2, we analyzed the individuals from the 67 European stands, together with the reference individuals (from the native range) from our 38 North-American reference stands, using the software STRUCTURE. We used a fixed K = 2 value to determine the assignment of each population and individual to one of the two varieties (coastal or Rocky Mountain). The purpose of this step was to detect cases of artificial intervarietal admixture in European stands. Next, we separated and removed pure Rocky Mountain Douglas-fir individuals (Q < 0.1) from introduced stands with affinity to coastal Douglas-fir (Q ≥ 0.9) and vice-versa.
After removing these individuals, we performed successive assignment analyses with GENECLASS2 following the hierarchical structure of the reference populations. We assigned the European populations to one of the two varieties at HL1. At this step, we used all 67 European Douglas-fir populations and assigned them to one of the two varieties. Next, we performed separate runs at the HL2 level within each of the two varieties. We employed the analyses at HL3 level, i.e. within each one of the clusters 1–4. The final step was to assign each of the 67 European populations to one of the 38 reference stands from North America by running the analysis within each one of clusters I–XII at HL4 according to the outline shown in Fig. 4.
The assignment tests resulted in a score for each assignment of a population to a given reference sample. This score (scorei,l) is defined as:
where Lij is the likelihood of an individual or a group of an individual i to belong to reference population l (Piry et al. 2004). The summary of the assignment analysis results at the population level are shown in Table 2. The result details for each of our 67 European Douglas-fir stands of unknown origin are presented in the Appendix (Online Resource 3). 64 of the 67 European populations (see Table 2) come from areas located in Washington, Oregon and Northern California (clusters I and II). Figure 5 shows the assigned location of the 64 European Douglas-fir stands to their closest reference population in North America. Next, we performed additional assignment analysis at the individual level to identify possible cluster-admixed stands. Cluster-admixed stands are introduced populations which might have been established with more than one seed source.

Population assignment of European stands of coastal Douglas-fir. Left, the reference populations of origin are marked with squares filled with different colors. Black dots symbolize reference populations without any assigned European populations. Right, the European populations are sorted according to the assignment. The colors correspond to the reference populations from North America
Performing only the assignment for groups of individuals, we can assign a population of unknown origin to one of the reference populations. Hence, a genetically intermediate population (cluster-admixed) population cannot be identified. For instance, at HL1, there are only two reference populations corresponding to the two varieties. Thus, it is not possible to characterize a European population as intermediate when performing ‘assignment of groups of individuals’ using GENECLASS2, because no such reference population exists. Thus, we additionally performed “assignment of individuals” and calculated the proportion of assigned individuals per cluster.
Assuming that no admixture is evident, the number of individuals assigned to the ‘true’ cluster will depend only on the accuracy of the method and will thus be similar to the CA value from the self-assignment (the measure of method accuracy). Deviations between the two values suggest an intermediate origin. We defined a tolerance level of 10% in the deviation from the CA score. Any higher deviation indicates that cluster-admixed stands may exist. The results of the assignment analysis at the individual level for identifying possible cluster-admixed populations are presented for each of our 67 European Douglas-fir stands in Online Resource 4.
Final results and discussion
Using the reference data (see Fig. 1) which covers the full natural distribution range of Douglas-fir in North-Western America, we are able to “trace back” and identify Central European Douglas-fir stands planted at the end of the 19th and early twentieth century of unknown seed origin (see Table 1). Note that compared to the provenance trials which were established during the 1960s across Europe (Chakraborty et al. 2015) the seed origin of the old European Douglas-fir stands were unknown, although some of these stands are used as European Douglas-fir seed sources.
At the hierarchical level 1 (HL1), we assigned only one population (S05) to the Rocky Mountain variety by both STRUCTURE and GENECLASS2 (see Online Resource 3). Further two populations (AZ06 and HO4) were assigned to the Rocky Mountain variety using GENECLASS2, but showed mixtures with the coastal variety based on the results of the Bayesian clustering analysis (see Online Resource 3). The seed sources of the remaining 64 of our 67 investigated Douglas-fir stands come from cluster I and II within the hierarchical level 3 (HL3) which refer to locations in Washington, Oregon and northern California (Table 2). Figure 5 shows a detailed map with the European Douglas-fir seed stands and their identified North American seed source location.
In Germany and Austria, many recommended provenances (see also the provenance trials established in the 1960) originate from cluster I (coastal variety), which covers an area between central Washington and the northern part of California (Konnert et al. 2008; Weißenbacher 2008). Most of our investigated old Douglas-fir stands of unknown origin came from the same area covering population R11 (Ashford/Elbe, Washington) with more than half of our 67 analysed stands (see Table 2), followed by R30 (Zone 061, Oregon) (see Table 2). Two reasons for this result seem to be possible: (1) a selection of former planted stands from unsuitable provenances, where only the “adapted” provenances (i.e. from the area of cluster I) survived; (2) a selection of stands with some early knowledge using seeds form cluster I. For example, reports about the excellent performance since more than a century are evident (see Schwappach 1911; Kanzow 1937) and our results confirm that the detected origin of our old Central European Douglas-fir stands are consistent the recommendations (Konnert et al. 2008; Weißenbacher 2008).
If Douglas fir stands regenerate naturally, the information about the genetic admixture enhances silvicultural decision making (Eckhart et al. 2017). For instance, stands from unsuitable provenances will contribute to the genepool of the regeneration of suitable stands because pollinations have a negative effect on the regeneration (Konnert and Ruetz 2006).
From our analysis, two important facts are of interest: (i) several stands originated from areas outside the recommended regions (e.g. two stands were assigned to a reference population from northern California), indicating that additional seed sources may be suitable for Douglas-fir in Europe and (ii) the distribution of the identified seed origins of our 67 Douglas-fir stands did not follow any geographic or climatic pattern within Europe.
Stands of the same origin occur for example in western Germany as well as in eastern Austria, climatically very distinct geographic zones. This suggests that the establishment of these stands did not follow a systematic pattern. It is probably a result of the high genetic diversity of populations within cluster I and II (van Loo et al. 2015) and the resulting adaptation potential. This indicates the important role of genetic diversity of seed sources for Douglas-fir management.
As expected, the geographic distribution of the clusters (see Fig. 3) follows a similar pattern as shown by van Loo et al. (2015). Only minor differences are evident due to the stochasticity of the applied Bayesian clustering method (Gilbert et al. 2012). Most of the 12 clusters at the HL3 are genetically and spatially delineated (see Figs. 2, 3, Online Resource 2). For instance, in Cluster 2 all four populations were assigned to one separate cluster (clusters III, IV, V and VI) at a membership proportion greater 0.9 (Q > 0.9; see Fig. 2).
Within HL3, cluster I and II exhibit a significant differentiation (see Online Resource 2) with a varying gradient in the membership proportion (Q-values). In Cluster I, the membership proportion decreased from south to north, while in Cluster II, it increased from south to north (see Fig. 2). Similar effects in densely sampled areas have been reported in other studies (e.g. Ekins et al. 2006) and may be seen as an effect of isolation by distance. We addressed this problem by introducing intermediate populations (0.4 < Q < 0.6) e.g. R16 (Reference population 16) as separate reference populations in our assignment performance (see Figs. 2, 3, 4). This setting also influences the test for detecting cluster-admixed stands: while at the HL1 and HL2, only seven potentially cluster-admixed stands were identified, at HL3 16 potentially admixed Douglas-fir stands between the clusters I and II (see Online Resource 4) were registered. This suggests that results have to be interpreted carefully when clusters represent areas located in the mating contact zone.
Cluster I and II are located within the most important area of seed sources for Central Europe (Konnert et al. 2008; Weißenbacher 2008) and would not be detected without performing the hierarchical step by step analysis of the reference dataset (see Figs. 2, 3). This method is a powerful approach to analyse populations with varying sampling density among clusters or hierarchical phylogenetic structure within clusters (Neophytou 2014; Janes et al. 2017; Wang 2017).
While the forest areas in Oregon and Washington were intensively sampled, we only had a few reference populations from the North (British Columbia) and the South (California) in our reference dataset (see Fig. 1). In such a situation, there is a tendency to assign individuals to a common cluster even if they are phylogenetically different (Kalinowski 2011; Neophytou 2014). Indeed, pairwise FSTs between British Columbian and Californian populations are high (see Online Resource 2) and do not support any common origin. Following our hierarchical approach, we were able to subdivide these two regions at the hierarchical level 3 (HL3; see Figs. 2, 3).
The accuracy assessment of our developed clustering approach based on a “leave-one-out-approach” method (see Fig. 4) revealed the highest consistency at the HL1 and HL2 (Hierarchal Level 1 and 2) with > 93.8% correctly assigned (CA) individuals to their clusters (variety) and a quality index (QI) of > 86.1% (see Fig. 4). At HL3 (Hierarchical Level 3), the clusters 2 and 3 (see Fig. 2), exhibit a CA of > 94% which strongly supports that seeds from Douglas-fir populations in California (clusters III, IV and V) or from British Columbia (cluster VI) are correctly classified. At the same Hierarchical Level, the clusters 1 and 4 exhibit a correct assignment (CA) between 75.3 and 79% and QI (Quality index ranging between 68.9 and 78.8% (see Fig. 2). This suggests that if Douglas-fir stands originate either from (i) the area including the northern Cascades and the southern part of Vancouver Island or (ii) the area between Southern Washington and Northern California, they have a 75.3% chance to be classified correctly. This is a result of (i) the low level of genetic differentiation and (ii) isolation by distance, which makes clustering difficult, as confirmed in previous findings with other tree species (Jolivet and Degen 2012; Degen et al. 2013; Nazareno and Dos Reis 2014).
At the lowermost hierarchical level, i.e. among single populations within a cluster from HL3, uncertainty of assignment increased. The best proportion of correctly assigned individuals (CA) was evident for cluster VII with 90%, while CA did not exceed 51.8% within clusters II, X and XI. Within the largest cluster I, only 33.1% of all individuals were assigned to their own population (see Fig. 2).
In addition to genetic differentiation, further factors affecting the accuracy of assignment are: (i) the number of analysed loci (in our study we used 13 high variable microsatellite loci; see Online Resource 1), (ii) their diagnostic power and (iii) the size of the reference populations (Cornuet et al. 1999; Bjørnstad and Røed 2002; Wang et al. 2009).
Conclusion
The selection of tree species, but also the most suitable provenance, is one of the key silvicultural decisions. Douglas-fir is considered as an important adaptation option for climate change because Douglas-fir exhibits a lower vulnerability if longer drought periods during the summer months occur versus our main native European tree species, e.g. Norway spruce or common beech. With an increase in temperature and changes in precipitation pattern (longer drought periods during the summer months) our native tree species are increasingly vulnerable in some regions, since they require a continuous water supply (e.g. rain) during the growth period. Our results are important for Douglas-fir management, because the investigated stands (i) have survived a full rotation length, (ii) show an excellent growth performance, (iii) regenerate naturally and some are (iv) identified and used as basic material for the production of reproductive material under the selected category. In addition, our assignment method can be used for other important questions associated to a changing climate such as (1) where do other European Douglas-fir stands come from? (2) Do all European stands come from a distinct area of North America or is the source geographically scattered? (3) Do established European stands follow the national provenance recommendation or do outliers exist? (4) Is there a correlation between side conditions within Europe and the origin of old Douglas-fir stands or not? (5) Do distinct provenances grow on special side conditions?
References
Allen GS (1960) A method of distinguishing Coastal from Interior Douglas Fir seed. Faculty of Forestry Research Note No. 28
Applied Biosystems (1998a) GeneScan analysis software user manual. Release 3.1. Applied Biosystems, Foster City, CA
Applied Biosystems (1998b) Genotyper 3.1. Applied Biosystems, Foster City, CA
Barner H (1973) Procurement of Douglas fir seed for provenance research. In: Proceedings IUFRO Meeting Working Party on Douglas-fir provenances. Göttingen, West Germany, pp 82–89
Bastien JCH, Sanchez L, Michaud D (2013) Douglas-Fir (Pseudotsuga menziesii (Mirb.) Franco). In: Pâques L (ed) Forest tree breeding in Europe: current state-of-the-art and perspectives. Springer, Dordrecht, pp 325–373
Baudouin L, Piry S, Cornuet JM (2004) Analytical Bayesian approach for assigning individuals to populations. J Hered 95(3):217–224
Bjørnstad G, Røed KH (2002) Evaluation of factors affecting individual assignment precision using microsatellite data from horse breeds and simulated breed crosses. Anim Genet 33(4):264–270
Breidenstein J, Bastien JC, Roman-Amat B (1990) Douglas-fir range-wide variation results from the IUFRO data base. In: Proceedings of the joint meeting of the western forest genetics association and IUFRO WorkingParties S202–05, 06, 12, and 14, Douglas-fir, Contorta Pine, Sitka Spruce, and Abies Breeding and Genetic Resources, Olympia, WA, USA, 20–24 August, pp 213–226
Campbell RK (1991) Soils, seed-zone maps, and physiography: guidelines for seed transfer of Douglas-fir in Southwestern Oregon. Forest Sci 37:973–986
Chakraborty D, Wang T, Andre K, Konnert M, Lexer MJ, Matulla C, Schueler S (2015). Selecting populations for non-analogous climate conditions using universal response functions: the case of Douglas-fir in central Europe. PloS one. https://doi.org/10.1371/journal.pone.0136357
Chakraborty D, Wang T, Andre K, Konnert M, Lexer MJ, Matulla C, Weißenbacher L, Schüler S (2016) Adapting Douglas-fir forestry in Central Europe: evaluation, application, and uncertainty analysis of a genetically based model. Eur J Forest Res 135:919–936
Chhatre VE, Emerson KJ (2017) StrAuto: automation and parallelization of STRUCTURE analysis. BMC Bioinformatics 18:192
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Darychuk N, Hawkins BJ, Stoehr M (2012) Trade-offs between growth and cold and drought hardiness in submaritime Douglas-fir. Can J For Res 42(8):1530–1541
Degen B, Ward SE, Lemes MR, Navarro C, Cavers S, Sebbenn AM (2013) Verifying the geographic origin of mahogany (Swietenia macrophylla King) with DNA-fingerprints. Forensic Sci Int Genet 7:55–62
Dunlap LH (1964) Differentiation of Coastal and Interior provenances using morphology of seed. MSc Thesis. University of British Columbia, Vancouver
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Eckert AJ, Bower AD, Wegrzyn JL, Pande B, Jermstad KD, Krutovsky KV, St. Clair JB, Neale DB, Pinaceae (2009) Association genetics of Coastal Douglas Fir (Pseudotsuga menziesii var. menziesii. I. Cold-hardiness related traits Genetics 182:1289–1302
Eckhart T, Walcher S, Hasenauer H, van Loo M (2017) Genetic diversity and adaptive traits of European versus American Douglas-fir seedlings. Eur J Forest Res 136(5–6):811–825
Eilmann B, de Vries SMG, den Ouden J, Mohren G, Sauren P, Sass-Klaassen (2013) Origin matters! Difference in drought tolerance and productivity of coastal Douglas-fir (Pseudotsuga menziesii (Mirb.)) provenances. For Ecol Manage 302:133–143
Ekins JE, Ekins JB, Layton L, Hutchison LA, Myres NM, Woodward SR (2006) Inference of ancestry: constructing hierarchical reference populations and assigning unknown individuals. Human Genomics 2(4):212
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578
Fussi B, Dounavi A, Konnert M (2013) Identification of varieties and gene flow in Douglas-fir exemplified in artificially established stands in Germany. Ann For Res 56:249–268
García D, Martínez A, Dunner S, Vega-Pla JL, Fernández C, Delgado JV, Cañón J (2006) Estimation of the genetic admixture composition of Iberian dry-cured ham samples using DNA multilocus genotypes. Meat Sci 72(3):560–566
Gilbert KJ, Andrew RL, Bock DG, Franklin MT, Kane NC, Moore JS, Brook TM, Renaut S, Rennison DJ, Veen T, Vines TH (2012) Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program structure. Mol Ecol 21(20):4925–4930
Gugger PF, Sugita S (2010) Glacial populations and postglacial migration of Douglas-fir based on fossil pollen and macrofossil evidence. Quat Sci Rev 29:2052–2070
Gugger PF, Sugita S, Cavender-Bares J (2010) Phylogeography of Douglas-fir based on mitochondrial and chloroplast DNA sequences: testing hypotheses from the fossil record. Mol Ecol 19:1877–1897
Hermann RK, Lavender DP (1990) Douglas-fir. In: Burns RM, Honkala BH (eds) Silvics of North America, vol 1. Conifers. USDA Forest Service, Washington, pp 527–540 (Handbook 654)
Holubčík M (1968) Cudzokrajné dreviny v lesnom hospodárstve. Slovenské vydavateľstvo pôdohospodárskej literatúry, Bratislava, p 371
Holubčík M (1974) Introdukcia cudzokrajných drevín-založenie a hodnotenie provenienčných pokusov s douglaskou. ZS. Zvolen, VÚLH, p 74
Holubčík M (1980) Introdukcia duglasky–hodnotenie proveniencií duglasky z hľadiska produkcie, kvality a odolnosti (Záverečná správa), Zvolen, p 48
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
IPCC (2007) Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Climate Change 2007: Synthesis Report (Pachauri RK, Reisinger A (eds)), Geneva, p 104
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K = 2 conundrum. Mol Ecol 24(14):3594–3602
Jolivet C, Degen B (2012) Use of DNA fingerprints to control the origin of sapelli timber (Entandrophragma cylindricum) at the forest concession level in Cameroon. Forensic Sci Int Genet 6:487–493
Kalinowski ST (2011) The computer program STRUCTURE does not reliably identify the main genetic clusters within species: simulations and implications for human population structure. Heredity 106(4):625
Kanzow H (1937) Die Douglasie: Aufstellung einer Ertragstafel auf Grund der Ergebnisse der preußischen Probeflächen und Auswertungen von Provenienzversuchen. Zeitschrift für Forst- Jagdwesen 69:65–93, 113–139, 241–271
Kleinschmit J, Bastien JC (1992) IUFRO’s role in Douglas-Fir (Pseudotsuga menziesii [Mirb.] Franco) tree improvement. Silvae Genetica 41:161–173
Klumpp R, (1999) Untersuchungen zur Genökologie der Douglasie (Pseudotsuga menziesii [Mirb.] Franco). Dissertation, Universität Göttingen
Köble R, Seufert G (2001) Novel maps for forest tree species in Europe. In: 8th European Symposium on the Physico-Chemical Behaviour of Air Pollutants: “A Changing Atmosphere!”, Turin 17–20 September 2001, pp 2–7
Kohl A, Nather J (1991) Die Douglasie. Kooperationsabkommen Forst–Platte–Papier, pp 12–15
Konnert M (2009) Genetische Aspekte und Herkunftsfragen bei Douglasie. Eberswalder forstliche Schriftenreihe. Band 43:28–32
Konnert M, Ruetz W (2006) Genetic aspects of artificial regeneration of Douglas-fir (Pseudotsuga menziesii) in Bavaria. Eur J Forest Res 125:261–270
Konnert M, Ruetz W, Schrimer R, Konnert M (2008) Fragen zum forstlichen Vermehrungsgut bei Douglasie. LWF Wissen 59:22–26
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191
Krutovsky KV, St. Clair JB, Saich R, Hipkins VD, Neale DB (2009) Estimation of population structure in coastal Douglas-fir [Pseudotsuga menziesii (Mirb.) Franco var. menziesii] using allozyme and microsatellite markers. Tree Genet Genomes 5:641–658
Lavender DP, Overton WS (1972) Thermoperiods and soil temperatures as they affect growth and dormancy of Douglas-fir seedlings of different geographic origin. Oregon State University, Corvallis
Li P, Adams WT (1989) Range-wide patterns of allozyme variation in Douglas-fir (Pseudotsuga menziesii). Can J For Res 19:149–161
Lowe J, Cross HB (2011) The application of DNA methods to timber tracking and origin verification. IAWA Journal 32(2):251–262
Martinsson O (1990) Research on Douglas-fir in Sweden. In: Proceedings Joint Meeting Western For. Gen. Ass. and IUFRO Working Parties Olympia, Washington, 4, 85:6
Matschiner M, Salzburger W (2009) TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics 25(15):1982–1983
Nazareno AG, Dos Reis MS (2014) Where did they come from? Genetic diversity and forensic investigation of the threatened palm species Butia eriospatha. Conserv Genet 15(2):441–452
Neophytou C (2014) Bayesian clustering analyses for genetic assignment and study of hybridization in oaks: effects of asymmetric phylogenies and asymmetric sampling schemes. Tree Genet Genomes 10:273–285
Neophytou C, Weisser AM, Landwehr D et al (2016) Assessing the relationship between height growth and molecular genetic variation in Douglas-fir (Pseudotsuga menziesii) provenances. Eur J Forest Res 135:465–481
Paetkau D, Slade R, Burden M, Estoup A (2004) Direct, real-time estimation of migration rate using assignment methods: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Pharis RP, Ferrell WK (1966) Differences in drought resistance between coastal and inland sources of Douglas fir. Can J Bot 44(12):1651–1659
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first-generation migrant detection. J Heredit 95:536–539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen X, Falush D (2010) Documentation for STRUCTURE software: version 2.3. Available with the program at http://pritch.bsd.uchicago.edu/structure.html
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. In: Proceedings of the National Academy of Sciences USA, 94, pp 9197–9201
Rau HM (2009) Bestände und Samenplantagen bei Douglasie. AFZ-Der Wald 5:220–221
Rehfeldt GE (1979) Ecological adaptations in Douglas-fir (Pseudotsuga menziesii var glauca) populations I. North Idaho and north-east Washington. Heredity 43:383–397
Rehfeldt GE (1983) Ecological adaptations in Douglas-fir (Pseudotsuga menziesii var. glauca) populations. III. Central Idaho. Can J Forest Res 13:626–632
Rosenberg N (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Schwappach A (1911) Die weitere Entwicklung der Versuche mit fremdländischen Holzarten in Preußen. Mitteilungen der Dtsch Dendrol Gesellschaft 20:3–37
Slavov GT, Howe GT, Yakovlev I, Edwards KJ, Krutovskii KV, Tuskan GA, Carlson JE, Strauss SH, Adams WT (2004) Highly variable SSR markers in Douglas-fir: Mendelian inheritance and map locations. Theor Appl Genet 108:873–880
Stephan BR (1980) Testing Douglas-fir provenances for resistance to Rhabdocline pseudotsugae by infection trials. Eur J For Pathol 20:152–161
Tigerstedt P (1990) Adaptability of seed sources across geographic zones—90 years of experiments in Finland. In: Proceedings Joint Meeting Western For. Gen. Ass. and IUFRO Working Parties. Olympia, Washington, 4, 85, 11p
Vähä JP, Erkinaro J, Niemelä E, Primmer CR (2007) Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol 16(13):2638–2654
van Loo M, Hintsteiner W, Pötzelsberger E, Schüler S, Hasenauer H (2015) Intervarietal and intravarietal genetic structure in Douglas-fir: nuclear SSRs bring novel insights into past population demographic processes, phylogeography, and intervarietal hybridization. Ecol Evol 5:1802–1817
Wang J (2017) The computer program STRUCTURE for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17(5):981–990
Wang J, Dobrowolski MP, Cogan NO, Forster JW, Smith KF (2009) Assignment of individual genotypes to specific forage cultivars of perennial ryegrass based on SSR markers. Crop Sci 49(1):49–58
Wei XX, Beaulieu J, Damase PK, Vargas-Hernández J, López-Upton J, Jaquish B, Bousquet J (2011) Range-wide chloroplast and mitochondrial DNA imprints reveal multiple lineages and complex biogeographic history for Douglas-fir. Tree Genet Genom 7:1025–1040
Weißenbacher L (2008) Herkunftswahl bei der Douglasie - der Schlüssel für einen erfolgreichen Anbau. BFW Praxis Inf 16:3–5
Acknowledgements
Open access funding provided by Austrian Science Fund (FWF). For financial support, we want to thank the following institutions, enterprises, state and private landowners: the Austrian Science Fund (FWF) (Project-ID: P26504), the Austrian Research Promotion Agency (FFG), the alps GmbH (Project: B04 AdaptAF B), the Austrian Chamber of Agriculture, the Chamber of Agriculture of Lower Austria, Austrian Federal Forests, Land and Forstbetriebe Niederoesterreich, the Metternich’sche Forstamt Grafenegg, the Forstamt Stift Goettweig, the Esterhazy Betriebe GmbH, the LIECO GmbH, the Montecuccolli Gut Mitterau, the Fuerst Star- hemberg‘sche Familienstiftung, the Gutsverwaltung Bubna-Litic, the Hatschek Forste, the Hoyos‘sche Forstverwaltung Horn, the Forstamt Ottenstein for financial support. Also, we would like to thank Lambert Weissenbacher from the Federal Research and Training Centre for Forests, Natural Hazards and Landscape for his help with the sample collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Christian Ammer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hintsteiner, W.J., van Loo, M., Neophytou, C. et al. The geographic origin of old Douglas-fir stands growing in Central Europe. Eur J Forest Res 137, 447–461 (2018). https://doi.org/10.1007/s10342-018-1115-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-018-1115-2