
Abstract
The yellow-legged hornet (Vespa velutina) is a social Hymenoptera native from Asia and an invasive species in Europe, where it was first detected in France in 2004. Since then, the species has spread across the continent invading mainland Spain and Mallorca island (Balearic archipelago, Western Mediterranean) in 2010 and 2015, respectively. Yellow-legged hornets cause severe damage to ecosystem by predating over a wide variety of pollinators including honeybees. Such a threat situation requires the development of effective management and prevention plans, which can greatly benefit from knowing both the origin and the genetic structure of the invading populations. Here we conduct a genetic study to shed light on both the origin and the phylogenetic relationships of V. velutina populations from Mallorca and mainland Spain using nuclear (STRs) and mitochondrial (cytochrome oxidase c subunit 1) gene markers. Our results show that Mallorca populations originated from invasive European specimens. Moreover, FST values, DAPC and genetic structure analysis suggest two independent incursions in the island with bottleneck and founder effect signatures. Finally, we contribute additional genetic evidence of the polyandrous behavior of this invasive species based on the inference of a mean number of mattings per nest of 3.94 (range 2–6.5). This study supports the human-mediated pathways of this species and highlights the importance of implementing effective biosecurity measures to prevent the spread of invasive alien species in island habitats.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Key Message
-
We studied the population genetics structure of Vespa velutina in Mallorca.
-
We identified the same mitochondrial haplotype shared by other European populations.
-
Two independent introductions from two different European regions were detected.
-
Identifying invasive populations pathways may help for invasive species management.
Introduction
Social insects have high invasion capacities (Beggs et al. 2011) mainly due to the ability of single foundress to establish a huge colony and to produce many future queens which in turn offer a large dispersal capacity. Such colonization abilities can be further enhanced by global trade and human mobility (Mikheyev et al. 2009; Arca et al. 2015). The main ecological effects of invasions include the impact on biodiversity (Vilà et al. 2011) and ecosystem functioning (Pejchar and Mooney 2009), with island environments being more vulnerable than continental systems due to their lower complexity, high endemicity rates and less diversity levels (Traveset and Richardson 2006, 2014; Sax and Gaines 2009).
The yellow-legged hornet (Vespa velutina) displays unique biological and life history traits that increase the probability of success in establishing and spreading in new territories (Moller 1996), including excellent dispersal capacities, high reproductive rates, broad diets and habitat ranges, effective predator defenses, and superior competitive skills (Moller 1996; Beggs et al. 2011). Consistently, this species is known to be the first invasive Vespidae predator accidentally introduced from Asia to Europe (Monceau et al. 2014). It was detected in France in 2004 from where the species has successfully spread and stablished in neighboring countries (Laurino et al. 2020). It was first reported from Spain in 2010 (López et al. 2011) where their populations have colonized all north Iberian peninsula arriving to Mallorca island (Balearic Islands, Western Mediterranean, Spain) in 2015 (Leza et al. 2021) probably due to an accidental introduction by humans (Robinet et al. 2019).
The first report of V. velutina in Mallorca leaded to the implementation of an intensive control protocol that allowed the removal of 31 nests and the capture of 16 queens using spring trapping between 2015 and 2018 (Leza et al. 2021). After these interventions, no more nests were found and the species was declared officially eradicated in November 2020 (CAIB 2020) until July 2021 when a new secondary nest was detected and removed in the island. Since then, neither adults nor nest have been detected in the archipelago.
While there is a wealth of studies on different aspects of the biology, ecology and management of V. velutina in Europe, only a few of them have focused on the genetics of the invasion (Arca et al. 2015; Budge et al. 2017; Granato et al. 2019; Jones et al. 2020; Quaresma et al. 2022). These studies have evidenced that the origin of the European invasion by yellow-legged hornets represented a genetic bottleneck event that explains their low levels of genetic diversity, probably as the result of a single queen introduction from southeast China (Arca et al. 2015). Nonetheless, this has not meant any limitation for the species to expand throughout the European continent. Such invasive capacity was also observed in Mallorca, where both the number of reported nests and the colonized area gradually increased since its detection until its eradication, after a potential population bottleneck event (Leza et al. 2021).
In this study, we expand the current knowledge on the invasion genetics of V. velutina in Europe based on mitochondrial (cox1) and nuclear (STRs) DNA analyses of the invading populations in the westernmost Mediterranean archipelago.
Materials and methods
Sampling and DNA extraction
A total of 335 adults of V. velutina were collected in Spain: Asturias (N = 11), Basque Country (N = 20), Catalonia (N = 16), Extremadura (N = 2), Galicia (N = 12) and Mallorca (N = 274) (Fig. 1, Table S1). Among sampled populations, only two individuals could be obtained from Extremadura (Spain) being the first detection of this invasive species in Extremadura. This particular sample was however retained in the analyses to increase the coverage of hornet invasive range. Queen individuals were collected from traps, workers were obtained from both traps and nests, while males were obtained directly from nests. Samples were stored in absolute ethanol and/or at − 20ºC until molecular analysis.

Maps of the Iberian Peninsula and Mallorca showing the location of the Vespa velutina individuals sampled from nests (black dots) and from traps (red dots), respectively. Pink-shaded areas indicate the invaded territories in Spain according to Laurino et al. (2020). Maps were obtained with Google Maps (Map data 2020, Google) using the function ‘get_map’ in the package ‘ggmap’ version 3.0.0.902 in R version 3.6.3
Genomic DNA was individually extracted and purified from insect legs using the Qiagen DNeasy Blood & Tissue kit (Qiagen) following the manufacturer’s instructions and RNA was removed using 60 μg of RNAse A (Promega).
Mitochondrial DNA sequencing and analysis
Genetic analyses based on mitochondrial cytochrome c oxidase subunit 1 (cox1) gene sequences were conducted on a sub-sample of 103 V. velutina individuals from Asturias (n = 10), Basque Country (n = 15), Catalonia (n = 15), Extremadura (n = 2), Galicia (n = 12), and Mallorca (n = 49). Such analyses included all individuals from mainland Spain, at least one individual from each nest detected in Mallorca and all trapped queens. Therefore, we cover all potential cox1 diversity present on the island. For each specimen, a cox1 gene fragment was PCR-amplified and sequenced using a custom designed V. velutina specific primer-pair Vespa_LCO (ATTCAACAAATCACAAAGATATTGG)/Vespa_HCO (TAAACTTCTGGATGTCCAAAGAATCA) targeting the standard DNA barcoding 658 bp region. Primers were designed based on available V. velutina complete mitochondrial genome sequences in GenBank (accession codes AP017943, AP018460, AP018461, AP018483, AP018484 and KY091645). PCR conditions used 0.2 μM of each primer and 3.5 mM MgCl2 (50 mM) using a standard protocol with an initial denaturation stage at 94 ºC (4 min), followed by 40 cycles with annealing temperature at 48 ºC (30 s), denaturation at 94 °C (30 s) and elongation at 72 °C (60 s), and a final extension step of 10 min. at 72 ºC. PCR products were checked for amplification by electrophoresis on 1% agarose gel stained with ethidium bromide and subsequently purified using the MSB Spin PCRapace kit (Invitek). Sanger sequencing was performed with the same primers as above using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems). Electropherograms were assembled and edited with CodonCode Aligner (CodonCode Corporation) and their respective identity as V. velutina cox1 sequences was assessed by comparing them with those available in GenBank using the BLASTn online tool (https://blast.ncbi.nlm.nih.gov/).
Newly generated cox1 sequences were aligned using MAFFT 7 (Katoh et al. 2018) under default options with all available V. velutina cox1 sequences in GenBank. A median joining haplotype network was computed using the popART 1.7 software (Leigh and Bryant 2015). In addition, the same DNA alignment was also used to infer a maximum likelihood (ML) phylogenetic tree in IQ-TREE v 1.6.12 (Nguyen et al. 2015) by simultaneously searching for the best evolutionary model according to Bayesian Information Criterion (TN + F + G4) and specifying 1000 bootstrap replicates. The resulting tree was visualized with FigTree v1.4.4 (Rambaut 2018).
STR amplification and analysis
All sampled individuals (335) were genotyped for 15 microsatellite loci using oligo combinations developed by previous studies on V. velutina population genetics (Daly et al. 2002; Hasegawa and Takahashi 2002; Arca et al. 2012). Forward primers were labeled with 6-FAM (blue), VIC (green), NED (yellow) and PET (red) dyes and the PCR amplifications were segregated into two multiplex reactions (Table S2). PCR reactions were performed in a total volume of 10 µL including 1 µL of V. velutina template DNA, 5 µL of Multiplex PCR kit (Qiagen), 3.6 µL of primer mix (10 µM) and 0.4 µL of H2O. The PCR conditions consisted of an initial denaturation step at 95 ºC (15 min) followed by 40 cycles at 94 ºC (30 s), 50 ºC (90 s) and 72 ºC (60 s), followed by a final extension stage at 60 ºC (30 min). Fragment analysis was performed on an ABI Prism 3130 DNA Genetic Analyzer (Applied Biosystems, Foster, California, USA), using GeneScan 500LIZ® as internal size standard. Fragment lengths were determined using GENEMAPPER 5.0 (Applied Biosystems) and checked manually.
The microsatellite dataset generated from the 335 V. velutina individuals was merged with an existing dataset containing genotypes from 417 individuals collected in other invaded territories (mainland Spain: Basque Country = 3 and Galicia = 42, France = 83, Italy = 11, Portugal = 191, and Korea = 8) and also from the native range of the species (Chinese provinces of Yunnan = 20 and Zhejiang/Jiangsu = 30, Indonesia = 21, and Vietnam = 8) (Arca et al. 2015; Quaresma et al. 2022). To enable dataset merging, allele scores of each microsatellite locus were harmonized between laboratories by genotyping 10 DNA samples used by Quaresma et al. (2022), which in turn were previously harmonized with Arca et al. (2015). Although the studies by Budge et al. (2017) and Jones et al. (2020) also generated V. velutina STR data, we opted for the conservative approach of not including them in our analyses since they were not harmonized with other studies.
Observed number of alleles (Na), observed heterozygosity (Ho) and expected heterozygosity (He) were computed per locus and region using ARLEQUIN 3.5.2.2 (Excoffier et al. 2005); whereas, unbiased expected heterozygosity (uHe) was estimated using the formula uHe = He*(2*n_individuals/(2*n_individuals-1)) (Nei 1978) and allelic richness (Ar) was estimated using the R package pegas (Paradis 2010), using the rarefaction method described by Hulbert (1971). In addition, inbreeding coefficient (f) was calculated using the formula f = (uHe-Ho)/uHe, where positive values represent an excess of homozygotes and negative values represent excess of heterozygotes (Li and Horvitz 1953). For each population, the mean and standard deviation of the above-mentioned genetic parameters were calculated to enable comparisons. Genetic differentiation among populations from the invaded and native ranges was estimated based on the pairwise FST values computed in ARLEQUIN 3.5.2.2 and using 10,000 random permutations to assess significance.
Population structure and relationships
The population structure was analyzed using the fast maximum likelihood genetic clustering approach (Beugin et al. 2018). This approach is very similar to the model implemented by STRUCTURE (Pritchard et al. 2000) but allows for much faster estimation of genetic clusters thanks to the use of the Expectation-Maximization (EM) algorithm. We initially investigated the number of clusters by using the k-means algorithm, where the preferred number of clusters was evaluated using the Bayesian information criterion (BIC) score.
A discriminant analysis of principal components (DAPC) was also used to infer population structure, using the adegenet 2.1.6 R package (Jombart 2008). This multivariate analysis combines principal component analysis (PCA) and discriminant analysis to determine the number of genetic clusters in the sample.
The program GeneClass v.2.0 (Piry et al. 2004) was used to assign or to exclude reference populations from either the invaded or the native ranges as possible sources of the Mallorca individuals on the basis of multilocus genotypes by using the standard criterion described by Rannala and Mountain (1997). The Monte Carlo resampling method (Paetkau et al. 2004) was also applied to identify the accurate critical values of exclusion/inclusion by simulating 10,000 genotypes for each population with a threshold probability value of 0.05.
Bottleneck detection
The occurrence of recent genetic bottlenecks in Mallorca populations was explored with the test of heterozygosity excess implemented in BOTTLENECK 1.2.0.2 (Cornuet and Luikart 1996; Piry et al. 1999). Heterozygosity excess is expected in populations that have experienced a significant reduction in size because rare alleles are lost (Cornuet and Luikart 1996). Two mutation models were tested: the infinite allele model (IAM) and the two-phase model (TPM), the latter of which incorporates elements of the IAM and stepwise mutation model (SMM) (variance = 12, SMM = 95%, Piry et al. 1999). The Wilcoxon sign-rank test was used to test for a statistically significant bottleneck (Luikart and Cornuet 1998).
Mating statistics
Both queen and mate genotypes were inferred from worker offspring genetic data using Colony 2.0.1.1 (Wang and Santure 2009) based on the 15 STR loci from Mallorca population. When available, males were also genotyped and known queen alleles were inferred since the haploid genotypes of males are the direct product of queen meiosis. Four trials with assumed genotyping error rates of 0.001, 0.01, 0.05, and 0.1 were performed. The allele frequencies inferred from Mallorca population between 2015 and 2021 were used as input.
Results
Mitochondrial DNA sequencing and analysis
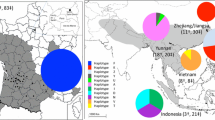
All specimens sampled from Mallorca and mainland Spain (Asturias, Basque Country, Catalonia, Extremadura and Galicia) shared the same cox1 haplotype (GenBank accession codes: ON094523 to ON094624), which was identical to the haplotype shared by all European V. velutina invaders sequenced to date and also by the specimens sampled in Jiangsu and Zhejiang (China) (Haplotype H1, Fig. 2). Moreover, our phylogenetic analysis also revealed a possible new haplotype from China (GenBank: MN716845) (Fig. 2). The possibility that the number of haplotypes is greater than that currently available in GenBank is consistent with the study by Perrard et al. (2014), which describe additional mtDNA haplotypes of V. velutina raising the total number to 25. However, these haplotypes are not available in GenBank and therefore have not been included in our analyses.

a Maximum Likelihood tree (log-likelihood = − 1942.5559) computed using the TN + F + G4 evolutionary model selected by the ModelFinder algorithm (Kalyaanamoorthy et al. 2017) implemented in the IQ-TREE software. Node numbers represent ultrafast bootstrap support (only values > 75 are shown). Samples contributed by this study are marked with asterisk symbols. b Median joining network of V. velutina cox1 haplotypes inferred in PopART. The pie charts display haplotype frequencies in each sampled region with size proportional to the number of sequences included (see scale at the bottom left). Missing haplotypes and mutational steps are indicated with solid black circles and hatch marks, respectively
The phylogenetic analyses based on cox1 sequences showed V. velutina divided into two main clades (Fig. 2a) in agreement with previous reports (Perrard et al. 2014; Takeuchi et al. 2017; Granato et al. 2019). The inferred tree supported the results derived from the network analysis (Fig. 2b) and showed the Spanish samples nested within the European invasion clade.
STR amplification and analysis
The mean (± SE) number of alleles (Na), allelic richness (Ar), and heterozygosities (Ho, uHe) computed for each sampled region of the invaded and native ranges in Europe and Asia are shown in Fig. 3.

Mean and standard error of number of alleles (Na), allelic richness (Ar), heterozygosities (Ho, uHe) and inbreeding coefficient (f) for each studied population from native and invaded range of V. velutina. Numbers following Mallorca codes refer to the year of collection (e.g., Mallorca 15 stands for samples collected in Mallorca in 2015)
The 15 microsatellite loci represented 180 different alleles, of which 174 were present in Asia and 64 in Europe. The number of private alleles was 116 and 6 in the native range and Europe, respectively. All loci from Asia were polymorphic, whereas in mainland Europe two loci were monomorphic (D2-142 in Portugal, mainland Spain, Italy and France, and R1-77 in mainland Spain), five loci were monomorphic in Mallorca between 2015 and 2018 (D2-142, D2-185, R1-77 and R1-169) and three loci were monomorphic in Mallorca 2021 (R1-80, D2.142 and R1-77).
Population structure and relationships
There was considerable divergence among populations: Pairwise FST ranged from 0.0001 to 0.6366 and all values were statistically significant (p value < 0.05), except between Mallorca 2015 and 2016 which was not different from zero (p value = 0.43) (Fig. 4). Generally, higher FST values corresponded to comparisons between native and invasive populations (excepting the pair Korea—Zhejiang/Jiangsu). Regarding the Mallorca populations, all pairwise comparisons between samples from 2015 to 2018 yielded low FST values, in contrast with the higher FST values retrieved from the analyses between them and Mallorca 2021 (range: 0.4378–0.465). According to FST values, the closest European population to the group conformed by Mallorca 2015–2018 was the Italian one (range: 0.1791–0.3392), while Mallorca 2021 was retrieved as closely related to V. velutina individuals from Catalonia (0.1822).

Multi-locus estimates of pairwise FST genetic distances between V. velutina populations from both invaded and native ranges (lower diagonal) and between Mallorca populations and Mainland Spain populations (top right). Numbers following Mallorca codes refer to the year of collection (e.g., Mallorca 15 stands for samples collected in Mallorca in 2015)
DAPC analyses clearly separated Asian and European populations (Fig. 5). Within the latter, the French populations were retrieved as the closest samples to the Asian ones (Fig. 5a). Consistent with FST estimates, Mallorca populations between 2015 and 2018 were located near to the Italian samples, and the Mallorca population 2021 near to V. velutina individuals from mainland Spain (Fig. 5b). The DAPC plot explained 93% of the total variance.

Scatterplot of the discriminant analysis of principal components (DAPC) of a Asian populations, France, and b magnified view of other European countries. Individuals genotypes are represented in dots, while populations are distinguished by colors and 95% inertia ellipses. The DAPC plot explained 93% of the total variance
A pattern of genetic heterogeneity within the European range was detected by the Maximum Likelihood genetic clustering using the expectation–maximization algorithm implemented in the adegenet package (Fig. 6). At K = 2, there was a clear separation between native and France populations from the rest of the invasive populations in Europe. Further partitioning of variation (K from 3 to 5) revealed different genetic structures in Europe (Fig. 6). K = 5 showed that the invasion of Mallorca between 2015 and 2018 had a differentiated genetic structure (mean Q-value ± SE = 99.99 ± 0.0006%) that partially includes the Italian population (mean Q-value ± SE = 40.66 ± 12.47%). In the other hand, the Mallorca population from 2021 (mean Q-value ± SE = 99.99 ± 0.0000007%) clustered with samples from mainland Spain (mean Q-value ± SE = 67.22 ± 4.42%). In addition, Portuguese (mean Q-value ± SE = 97.87 ± 1.16%), French (mean Q-value ± SE = 99.99 ± 0.00003%) and Asian populations (mean Q-value ± SE = 99.99 ± 0.00007%) conformed three distinct clusters. Likewise, DAPC analyses identified the Asian, French, Atlantic (Portugal + Spain), Mediterranean (Spain + Italy) and Mallorca as differentiated genetic entities (Fig. 5).

Genetic structure of V. velutina. Populations admixture ancestry components are shown from K = 2 to K = 5 for the 752 individuals. The lowest Bayesian information criterion (BIC) was obtained at K = 5. Populations are delimited by black borders
Notably, both DAPC (Fig. 5) and admixture (Fig. 6) analyses supported the genomic distinctiveness of Mallorca population between 2015 and 2018 from all other V. velutina populations.
One individual from Mallorca 2015 was detected by GENECLASS as first-generation migrant (p-value = 0.004) with a putative origin in Italy (log(L) = − 6.804). Likewise, an individual from Mallorca 2017 was also detected as first-generation migrant (p-value < 0.001) with a putative origin located in mainland Spain (log(L) = − 8.926).
Since FST values, DAPC and genetic structure analyses suggest two different introductions from mainland Europe in Mallorca, invasive populations sampled in the island were assigned by GENECLASS to two different regions: Mallorca samples between 2015 and 2018 were assigned to Italy (score = 100%), and those from 2021 were assigned to mainland Spain, specifically Catalonia (score = 100%).
Bottleneck detection
The decrease in genetic diversity observed in the invasive populations may be indicative of a bottleneck event (Arca et al. 2015). In this regard, the heterozygosity excess indicating a genetic bottleneck was tested in Mallorca populations between 2015 and 2021. The results were consistent with a bottleneck episode according to the significant value of heterozygosity excess estimated under both IAM and TPM models (p value < 0.05).
Mating statistics
Depending on the assumed genotyping error rate, the total inferred number of fathers varied from 1 to 10 over the Mallorca nests studied. On the one hand, all nest from 2015 to 2018 (first invasion) presented different patrilines resulting in a mean number of observed mattings per nest of 3.94 (range 2–6.5). On the other hand, a single patriline was detected in the only nest found in 2021 (second invasion). Diploid males were detected in two nests collected in 2017 and 2021, respectively.
Discussion
Our results are compatible with a scenario in which V. velutina populations in both mainland Spain and the Balearic archipelago could have derived from the spreading southward of the V. velutina population initially established in France, rather than from multiple independent introductions from the native range. This view is supported by the identification of a single mitochondrial haplotype shared by all our samples and those previously reported from the remainder of the invaded territories in Europe (Arca et al. 2015; Budge et al. 2017; Granato et al. 2019; Husemann et al. 2020; Jones et al. 2020; Quaresma et al. 2022). Furthermore, the European origin of the invasions of both mainland Spain and the Balearic archipelago is reinforced by nuclear STR data revealing the existence of a shared allelic pool and similar genetic diversity levels (Na and Ar) (Arca et al. 2015; Budge et al. 2017; Jones et al. 2020; Quaresma et al. 2022). Indeed, the FST values reported here indicate that Mallorca populations are closely related to representatives from Italy (samples Mallorca 2015–2018) and Catalonia (single nest found in Mallorca in 2021), whereas mainland Spain colonies show closer genetic affinities with those from Italy and Portugal.
The invasion of Mallorca Island by V. velutina is compatible with a bottleneck event. The estimated excess of observed heterozygosity (Ho) and reduced inbreeding coefficient (f) can lead to a decrease in terms of both adaptive potential and fitness of a founding population, therefore compromising its establishment and spread across new ranges (Willi et al. 2006). However, there exists evidence that V. velutina populations can establish successfully even after a severe genetic bottleneck resulting from the introduction of very few or even a single female (Arca et al. 2015). The case of the invasion of Mallorca reported here reinforce this view, since even after a potential second bottleneck the invasive population of V. velutina kept growing after the first detection despite eradication efforts (Leza et al. 2021). In fact, the increase in observed heterozygosity detected on consecutive populations between 2015 and 2018 could be explained by the control and eradication efforts applied in the island (Leza et al. 2021). The eradication plan focused on entire nest removal before male emergences, therefore preventing reproduction and lowering the genetic diversity levels of the population and increasing the bottleneck event and founder effect on the next generations.
Genetic diversity of founders is critical for the adaptative response to new environments (Lockwood et al. 2005; Drake and Lodge 2006). In this regard, the polyandrous mating system of V. velutina allowed the introduction of an important fraction of the genetic diversity of the native population into Europe (Arca et al. 2015). Here we contribute additional genetic evidence of the polyandrous behavior of this invasive species based on the inference of up to 10 potential males fecundating a single queen. High allelic diversity in the founding individuals coupled with other extraordinary life history traits common to social Hymenoptera (Moller 1996; Beggs et al. 2011) probably helped spread of this invasive species after a new bottleneck event in Mallorca. According to our genetic results and ecological niche models developed specifically for the island (Herrera et al. 2023), if an early detection and eradication plan would not have been implemented (Leza et al. 2021) the species probably would have colonized and stablished in part of the island, as predicted by Robinet et al. (2019).
Bottleneck events, inbreeding, limited gene flow and genetic drift reduce sex allele diversity (Darrouzet et al. 2015). As well as other Hymenoptera species, V. velutina sex is determined by a multi-allelic locus in which females are diploid (heterozygous genotype) and males are haploid (hemizygous genotype) (Heimpel and De Boer 2008). When allelic diversity is low at the sex locus, there is a higher chance that the queen will produce homozygous eggs, which will develop into diploid males (Darrouzet et al. 2015; Quaresma et al. 2022) that are usually sterile and do not contribute to colony tasks. In this study we report two nests hosting diploid males (7.69% of all sampled nests) after a bottleneck event. This value is considerably lower than the ratios reported by studies conducted in United Kingdom and France, where diploid males represented 75 and 48% of all nests genotyped, respectively (Darrouzet et al. 2015; Budge et al. 2017).
Our case study represents the first time that the yellow-legged hornet reaches a Mediterranean island after its arrival to Europe in 2004 (Monceau et al. 2014). In fact, the genetic results presented in this study shows that this species has reached Mallorca in two different moments from two different European regions, respectively: Italy (2015) and Mainland Spain (2021). Therefore, all nests identified and eliminated in Mallorca during the eradication efforts can be attributed to a single introduction that was initially detected in 2015. Fortunately, thanks to an observant citizen, the second introduction in 2021 was swiftly detected, enabling the competent administration to promptly locate the nest and sent it to the University of the Balearic Islands for subsequent analyses. Given that the nest hosted male specimens, two possible scenarios were confronted: There was already a reproductive caste established indicating that the species was already dispersing, or the male specimens were sterile. Each of these scenarios could have distinct implications for the management tasks associated with controlling this invasive species. Consequently, a genetic analysis was conducted on male hornets using 15 STRs microsatellite markers revealing that all specimens were diploid, and therefore sterile (Heimpel and De Boer 2008; Darrouzet et al. 2015). The employed methodology provided valuable insights into the potential dispersal of this species, enabling us to predict whether the competent administration should be prepared to detect and eradicate new nests in the upcoming years in the way described by (Leza et al. 2021).
Robinet et al. (2019) explored the human-mediated dispersal of V. velutina concluding that the Mediterranean islands could not be naturally colonized, and only an anthropogenic introduction could favor the colonization of such territories. Mallorca island is 176 km away from the mainland nearest point, so the most probably introduction could be through shipping traffic. The genetic structure analyses performed here support the pathways of this species and assign French populations to a cluster that is more closely related to Asian populations than to the rest of European populations, in contrast with other genetic studies (Arca et al. 2015; Budge et al. 2017; Quaresma et al. 2022). Interestingly, the individuals sampled in Mallorca clustered into a well differentiated position from the rest of European or Asian clusters, which is consistent with the bottleneck event inferred by our analyses. This result may indicate that the diversity loss of invasive populations of V. velutina may vary across geographic range as documented for numerous animal and plant invasions (Forsman 2014).
Our findings reveal the pathways used for this species to reach new isolated territories as the Mediterranean islands. Such a threat situation requires the development of effective management and prevention plans, which can greatly benefit from knowing both the origin and the genetic structure of the invading populations.
Author’s contributions
CH, ML and JAJ-R conceived the study; CH and ML provided Vespa velutina samples; CH, JF and JAJ-R designed the experiments; CH, JF and JAJ-R performed the experiments and analyzed the data; CH and JAJ-R performed phylogenetic analyses, analyzed the data and created the figures; CH wrote the original draft, and all authors commented and contributed to the final manuscript. All authors read and approved the final manuscript.
References
Arca M, Capdevielle-Dulac C, Villemant C et al (2012) Development of microsatellite markers for the yellow-legged Asian hornet, Vespa velutina, a major threat for European bees. Conserv Genet Resour 4:283–286. https://doi.org/10.1007/s12686-011-9525-1
Arca M, Mougel F, Guillemaud T et al (2015) Reconstructing the invasion and the demographic history of the yellow-legged hornet, Vespa velutina, in Europe. Biol Invasions 17:2357–2371. https://doi.org/10.1007/s10530-015-0880-9
Beggs JR, Brockerhoff EG, Corley JC et al (2011) Ecological effects and management of invasive alien Vespidae. Biocontrol 56:505–526. https://doi.org/10.1007/s10526-011-9389-z
Beugin MP, Gayet T, Pontier D et al (2018) A fast likelihood solution to the genetic clustering problem. Methods Ecol Evol 9:1006–1016. https://doi.org/10.1111/2041-210X.12968
Budge GE, Hodgetts J, Jones EP et al (2017) The invasion, provenance and diversity of Vespa velutina Lepeletier (Hymenoptera: Vespidae) in Great Britain. PLoS ONE 12:1–12. https://doi.org/10.1371/journal.pone.0185172
CAIB (2020) Las Illes Balears se convierten en el primer territorio europeo que consigue erradicar la avispa asiática. https://www.caib.es/pidip2front/jsp/es/ficha-convocatoria/strongstronglas-istrongstrongllesstrongstrong-balears-se-convierten-en-el-primer-territorio-europeo-que-consigue-erradicar-la-avispa-asiaacuteticastrongstrong. Accessed 8 June 2021
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014. https://doi.org/10.1093/genetics/144.4.2001
Daly D, Archer ME, Watts PC et al (2002) Polymorphic microsatellite loci for eusocial wasps (Hymenoptera: Vespidae). Mol Ecol Notes 2:273–275. https://doi.org/10.1046/j.1471-8278
Darrouzet E, Gévar J, Guignard Q, Aron S (2015) Production of early diploid males by European colonies of the invasive hornet Vespa velutina nigrithorax. PLoS ONE 10:1–9. https://doi.org/10.1371/journal.pone.0136680
Drake JM, Lodge DM (2006) Allee effects, propagule pressure and the probability of establishment: Risk analysis for biological invasions. Biol Invasions 8:365–375. https://doi.org/10.1007/s10530-004-8122-6
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinforma 1:117693430500100. https://doi.org/10.1177/117693430500100003
Forsman A (2014) Effects of genotypic and phenotypic variation on establishment are important for conservation, invasion, and infection biology. PNAS 111:302–307. https://doi.org/10.1073/pnas.1317745111
Granato A, Negrisolo E, Bonomi J et al (2019) Recent confirmation of a single haplotype in the Italian population of Vespa velutina. Biol Invasions 21:2811–2817. https://doi.org/10.1007/s10530-019-02051-4
Hasegawa E, Takahashi J (2002) Microsatellite loci for genetic research in the hornet Vespa mandarinia and related species. Mol Ecol Notes 2:306–308. https://doi.org/10.1046/j.1471-8278
Heimpel GE, De Boer JG (2008) Sex determination in the hymenoptera. Annu Rev Entomol 53:209–230. https://doi.org/10.1146/annurev.ento.53.103106.093441
Herrera C, Jurado-Rivera JA, Leza M (2023) Ensemble of small models as a tool for alien invasive species management planning: evaluation of Vespa velutina (Hymenoptera: Vespidae) under Mediterranean island conditions. J Pest Sci 96:359–371. https://doi.org/10.1007/s10340-022-01491-7
Hulbert SH (1971) The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577–586
Husemann M, Dey LS, Hawlitschek O (2020) Vespa velutina nigrithorax Lepeltier, 1836 from Hamburg (Northern Germany) shares the same COI haplotype with other European populations. J Hymenopt Res 79:111–115. https://doi.org/10.3897/JHR.79.57048
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jones EP, Conyers C, Tomkies V et al (2020) Managing incursions of Vespa velutina nigrithorax in the UK: an emerging threat to apiculture. Sci Rep 10:1–8. https://doi.org/10.1038/s41598-020-76690-2
Katoh K, Rozewicki J, Yamada KD (2018) MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166. https://doi.org/10.1093/bib/bbx108
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14(6):587–589. https://doi.org/10.1038/nmeth.4285
Laurino D, Lioy S, Carisio L et al (2020) Vespa velutina: An alien driver of honey bee colony losses. Diversity 12. https://doi.org/10.3390/D12010005
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Leza M, Herrera C, Picó G et al (2021) Six years of controlling the invasive species Vespa velutina in a Mediterranean island: The promising results of an eradication plan. Pest Manag Sci 77:2375–2384. https://doi.org/10.1002/ps.6264
Li CC, Horvitz DG (1953) Some methods of estimating the inbreeding coefficient. Am J Hum Genet 5:107–117
Lockwood JL, Cassey P, Blackburn T (2005) The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20:223–228. https://doi.org/10.1016/j.tree.2005.02.004
López S, González M, Goldarazena A (2011) Vespa velutina Lepeletier, 1836 (Hymenoptera: Vespidae): First records in Iberian Peninsula. EPPO Bull 41:439–441. https://doi.org/10.1111/j.1365-2338.2011.02513.x
Luikart G, Cornuet JM (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237. https://doi.org/10.1046/j.1523-1739.1998.96388.x
Mikheyev AS, Bresson S, Conant P (2009) Single-queen introductions characterize regional and local invasions by the facultatively clonal little fire ant Wasmannia auropunctata. Mol Ecol 18:2937–2944. https://doi.org/10.1111/j.1365-294X.2009.04213.x
Moller H (1996) Lessons for invasion theory from social insects. Biol Conserv 78:125–142. https://doi.org/10.1016/0006-3207(96)00022-5
Monceau K, Bonnard O, Thiéry D (2014) Vespa velutina: a new invasive predator of honeybees in Europe. J Pest Sci 87:1–16. https://doi.org/10.1007/s10340-013-0537-3
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590. https://doi.org/10.1093/genetics/89.3.583
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65. https://doi.org/10.1046/j.1365-294X.2004.02008.x
Paradis E (2010) Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420. https://doi.org/10.1093/bioinformatics/btp696
Pejchar L, Mooney HA (2009) Invasive species, ecosystem services and human well-being. Trends Ecol Evol 24:497–504. https://doi.org/10.1016/j.tree.2009.03.016
Perrard A, Arca M, Rome Q et al (2014) Geographic variation of melanisation patterns in a hornet species: genetic differences, climatic pressures or aposematic constraints? PLoS ONE 9:1–16. https://doi.org/10.1371/journal.pone.0094162
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Piry S, Alapetite A, Cornuet JM et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quaresma A, Henriques D, Godinho J et al (2022) Invasion genetics of the Asian hornet Vespa velutina nigrithorax in Southern Europe. Biol Invasions 24:1479–1494. https://doi.org/10.1007/s10530-022-02730-9
Rambaut A (2018) FigTree v1.4.4. http://tree.bio.ed.ac.uk/software/figtree/
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. PNAS 94:9197–9201. https://doi.org/10.1073/pnas.94.17.9197
Robinet C, Darrouzet E, Suppo C (2019) Spread modelling: a suitable tool to explore the role of human-mediated dispersal in the range expansion of the yellow-legged hornet in Europe. Int J Pest Manag 65:258–267. https://doi.org/10.1080/09670874.2018.1484529
Sax DF, Gaines SD (2009) Species invasions and extinction: the future of native biodiversity on islands. PNAS 105:11490–11497. https://doi.org/10.1073/pnas.0802290105
Takeuchi T, Takahashi R, Kiyoshi T et al (2017) The origin and genetic diversity of the yellow-legged hornet, Vespa velutina introduced in Japan. Insectes Soc 64:313–320. https://doi.org/10.1007/s00040-017-0545-z
Traveset A, Richardson DM (2006) Biological invasions as disruptors of plant reproductive mutualisms. Trends Ecol Evol 21:208–216. https://doi.org/10.1016/j.tree.2006.01.006
Traveset A, Richardson DM (2014) Mutualistic interactions and biological invasions. Annu Rev Ecol Evol Syst 45:89–113. https://doi.org/10.1146/annurev-ecolsys-120213-091857
Vilà M, Espinar JL, Hejda M et al (2011) Ecological impacts of invasive alien plants: a meta-analysis of their effects on species, communities and ecosystems. Ecol Lett 14:702–708. https://doi.org/10.1111/j.1461-0248.2011.01628.x
Wang J, Santure AW (2009) Parentage and sibship inference from multilocus genotype data under polygamy. Genetics 181:1579–1594. https://doi.org/10.1534/genetics.108.100214
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458. https://doi.org/10.1146/annurev.ecolsys.37.091305.110145
Acknowledgements
The authors would like to thank the collaborators who provided us the samples for this study: Servei de Protecció d’Espècies of the Balearic Government and COFIB (Balearic Islands), Jaume Cambra Sánchez and Antoni Armengol (Catalonia), Sandra Rojas Nossa (Galicia), Rosa Maria Alonso Rojas, Omaira de la Hera and Roberto Puch Pérez (Basque Country), José Manuel González and Judith Pérez (Asturias) and Eva Frontera (Extremadura). Moreover, we thanks to Alice Pinto from Instituto Politécnico de Bragança for providing us 10 DNA samples used in their study to harmonize microsatellite locus between laboratories. Finally, thanks to the support of the Biodiversity Foundation of the Ministry for the Ecological Transition and the Demographic Challenge.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work has been possible thanks to a FPI grant (FPI_014_2020) from the Conselleria d'Educació, Universitat i Recerca del Govern de les Illes Balears. This study has been done with the support of the Biodiversity Foundation of the Ministry for the Ecological Transition and the Demographic Challenge with the project titled “STOP Vespa velutina: descifrando de dónde ha llegado y cómo se dispersa para establecer mecanismos de gestión de esta especie exótica invasora que amenaza a las abejas”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no potential conflict of interest in relation to the study in this paper.
Additional information
Communicated by Nuria Agustí.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herrera, C., Ferragut, J.F., Leza, M. et al. Invasion genetics of the yellow-legged hornet Vespa velutina in the Westernmost Mediterranean archipelago. J Pest Sci 97, 645–656 (2024). https://doi.org/10.1007/s10340-023-01680-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-023-01680-y