
Abstract
Purpose
In neurogenic orthostatic hypotension, blood pressure falls when upright owing to impaired release of norepinephrine, leading to dizziness. Ampreloxetine, a selective norepinephrine reuptake inhibitor, increases circulating norepinephrine levels. This study explored the safety of ampreloxetine and its effect on blood pressure and symptoms in patients with neurogenic orthostatic hypotension.
Methods
A multicenter ascending-dose trial (range 1–20 mg, Part A) was followed by a 1 day, double-blind, randomized, placebo-controlled study (median dose 15 mg, Part B). Eligible patients then enrolled in a 20-week, open-label, steady-state extension phase (median dose 10 mg, Part C) followed by a 4-week withdrawal. Assessments included the Orthostatic Hypotension Symptom Assessment Scale (item 1), supine/seated/standing blood pressure, and safety.
Results
Thirty-four patients (age 66 ± 8 years, 22 men) were enrolled. Part A: The proportion of participants with a positive response (i.e., increase from baseline in seated systolic blood pressure of ≥ 10 mmHg) was greater with the 5 and 10 mg ampreloxetine doses than with placebo or other active ampreloxetine doses. Part B: Seated blood pressure increased 15.7 mmHg 4 h after ampreloxetine and decreased 14.2 mmHg after placebo [least squares mean difference (95% CI) 29.9 mmHg (7.6–52.3); P = 0.0112]. Part C: Symptoms of dizziness/lightheadedness improved 3.1 ± 3.0 points from baseline and standing systolic blood pressure increased 11 ± 12 mmHg. After 4 weeks of withdrawal, symptoms returned to pretreatment levels. The effect of ampreloxetine on supine blood pressure was minimal throughout treatment duration.
Conclusion
Ampreloxetine was well tolerated and improved orthostatic symptoms and seated/standing blood pressure with little change in supine blood pressure.
Trial registration
NCT02705755 (first posted March 10, 2016).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Orthostatic hypotension (OH) is defined as a fall in systolic blood pressure (BP) of at least 20 mmHg or a reduction in diastolic BP of at least 10 mmHg within 3 min of standing or head-up tilt [1]. Estimates suggest that 20% of patients who meet criteria for OH have neurogenic OH (nOH) due to neurodegenerative lesions affecting the efferent sympathetic neuronal pathway, blunted reflex norepinephrine (NE) release, and insufficient vasoconstriction on standing [1,2,3]. In severe cases, patients with nOH can develop symptoms of cerebral hypoperfusion, generalized weakness, and an overwhelming urge to sit down [4]. This can lead to disability, poor quality of life, syncope, and falls [5, 6].
nOH is common in patients with synucleinopathies—a group of overlapping neurodegenerative diseases with intracellular deposits of misfolded α-synuclein and cell loss at various levels within the sympathetic neuronal pathway [2, 7,8,9]. In Parkinson’s disease (PD) and pure autonomic failure (PAF), α-synuclein aggregates as Lewy bodies and neurites mostly in the peripheral, postganglionic, sympathetic neurons innervating the blood vessels, and the heart [2, 7, 9]. In multiple system atrophy (MSA), the autonomic neuronal pathology occurs with a more central pattern. Initial loss impacts oligodendroglial cells leading to death of the sympathetic, preganglionic neurons of the spinal cord and brainstem nuclei [9]. Regardless of the site of the lesion, these three phenotypes share the failure to appropriately release NE on standing and all may lead to severe symptomatic nOH [2, 10].
Available treatment options include volume expansion with fludrocortisone or short-acting pressor agents, such as the direct alpha-1 adrenergic agonist midodrine or the norepinephrine precursor droxidopa [10,11,12]. However, these strategies do not target the residual sympathetic nerve activity, require multiple daily dosing, have significant side effects, and can induce supine hypertension. Moreover, many patients do not adequately respond and remain severely disabled [6, 13,14,15].
Norepinephrine transporter (NET) inhibitors are a novel pharmacological approach being explored as a treatment for nOH in the autonomic synucleinopathies [16]. By blocking the reuptake of NE, they harness residual peripheral sympathetic vasoconstrictor tone by prolonging the effect of released NE from the remaining postganglionic sympathetic neurons [17]. Because the central sympatholytic mechanism of NET inhibitors is not seen in patients with synucleinopathies [18], blockade of the reuptake of NE in the postganglionic sympathetic neurons may increase BP and improve symptoms [16, 17, 19].
Ampreloxetine is a novel, investigational, long-acting NE reuptake inhibitor (NRI). In addition to ongoing randomized and well-controlled phase 3 placebo-controlled studies in symptomatic nOH, ampreloxetine has previously been evaluated in up to 499 individuals, including healthy volunteers and individuals with fibromyalgia or attention deficit hyperactivity disorder. Ampreloxetine was found to be generally safe and well tolerated across these populations. Pharmacokinetic (PK)/pharmacodynamic evaluations conducted in these individuals have shown that ampreloxetine has a half-life of 30–40 h and a preferential selectivity for the NET over the serotonin transporter, with > 75% NET occupancy at a dose of 10 mg QD [20]. A phase 2 trial to investigate the safety, exploratory efficacy, and durability of once-daily oral ampreloxetine in patients with nOH due to synucleinopathies was conducted. Ampreloxetine PK results from this study have been published [21].
Methods
Trial oversight and registration
The phase 2 trial was a three-part, multicenter study conducted at six sites in the US (Long Beach, CA; Farmington Hills, MI; Berlin, NJ; New York, NY; Nashville, TN; Dallas, TX). The studies were conducted between September 2017 and November 2018 in accordance with the Declaration of Helsinki. Local institutional review board approval was obtained, and all participants signed informed consent. All authors reviewed the data, which was analyzed by the sponsor, and the study was funded by Theravance Biopharma R&D, Inc. The study was listed on ClinicalTrials.gov (NCT02705755) and is reported here following the CONSORT statement for reporting of clinical trials.
Trial design
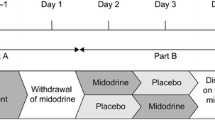
The trial (Fig. 1) began with a single-blind, 5-day, ascending-dose inpatient study (Part A). On day 1, placebo was administered, and assessments were performed by the unblinded investigator, but the participant was blinded. On days 2–5, participants were administered ascending oral doses of ampreloxetine (dried powder mixed with filtered apple juice prepared by an unblinded pharmacist) beginning at 1 mg, followed by 2.5, 5, and 10 mg or 2.5, 5, 10, and 20 mg, always at 8 a.m. after an overnight fast. Stopping criteria included safety concerns, intolerable side effects, seated systolic BP > 180 mmHg, or seated diastolic BP > 110 mmHg. Participants were discharged and underwent washout for a minimum of 8 days. Responders (i.e., patients with an increase of ≥ 10 mmHg in seated systolic BP relative to placebo) were eligible to participate in a double-blind, placebo-controlled, randomized study (Part B). Participants were randomly assigned 1:1 to ampreloxetine or placebo using a centralized computer-generated block randomization schedule. Based on pharmacokinetic modeling of ampreloxetine accumulation after escalating doses, participants received either 1.5 times their highest effective tolerated ampreloxetine dose (dried powder mixed with filtered apple juice prepared by an unblinded pharmacist) or matching placebo for 1 day with both the investigator and the participant blinded to treatment assignment. While inpatient, ampreloxetine was administered in the morning, and participants were given a standardized low-carbohydrate meal to avoid the confounding effects of postprandial hypotension and instructed to maintain a stable fluid intake to avoid volume shifts. The study was originally designed to only include Part A and B but was modified to include Part C (an open-label extension study) after several patients who had participated in Part A and/or Part B requested compassionate-use access to ampreloxetine based on their subjective experience of reduced nOH symptoms. As a result, the protocol was amended to terminate Part B enrollment early and to initiate the Part C open-label extension phase of the study. Participants in Part C underwent a washout for a minimum of 8 days after Part A or B before enrolling in Part C. To test durability, participants were readmitted to the hospital for 4 days to repeat pretreatment evaluations and restart ampreloxetine. The dose administered in Part C (day 1) was equal to 50% of the highest tolerated dose administered in Part A. The investigators could, however, double the dose at their discretion (up to day 29) or thereafter if the sponsor agreed. They were then discharged and instructed to take ampreloxetine capsules orally in the morning before breakfast for 20 weeks, which was then followed by a 4-week withdrawal phase to determine whether their symptoms worsened and returned to baseline. In the open-label, outpatient phase, ampreloxetine could be increased to a maximum of 20 mg once daily up to the end of week 4 with at least 7 days between dose increases. If the participant developed supine hypertension or other adverse events (AEs) ampreloxetine was stopped for 3 days and restarted at 50% of the previous dose. Amendments to the protocol included dropping the 1 mg dose in Part A to begin the dose escalation at 2.5 mg and early termination of Part B allowing for patients to directly enroll in the open-label extension phase (Part C).

Overall trial design. Part A: Day 1, placebo was administered single blind and on subsequent days, participants received ascending doses of ampreloxetine (1–10 mg or 2.5–20 mg from day 2 to day 5). Stopping criteria included safety concerns/adverse events or seated BP > 180/110 mmHg. Responders were defined as those with a seated systolic pressor response ≥ 10 mmHg relative to placebo and underwent a washout. Part B: Participants were randomized (1:1) to ampreloxetine or placebo for 1 day. Part A and Part B were inpatient studies and the primary endpoint in both was the change in seated BP 6–8 h post-ampreloxetine. Part C: Predose assessments were taken as baseline. Participants received open-label ampreloxetine for 20 weeks as outpatients (median dose 10 mg/day). The primary endpoint in Part C was a change from baseline in OHSA #1 scores at week 4. At the end of the 20 weeks, ampreloxetine was withdrawn to determine whether symptoms diminished to baseline levels. Key: Blue pills = ampreloxetine. Gray pills = placebo. Red = primary endpoints for Parts A, B, and C. Green asterisk = main protocol amendments (i) to start the dose escalation at 2.5 mg and (ii) to eliminate Part B in order for patients to directly enter Part C. BP blood pressure, h hours, mg milligrams, min minutes, mmHg millimeters of mercury, OHSA #1 dizziness/lightheadedness score on Orthostatic Hypotension Symptom Assessment (question 1), R randomization, SBP systolic blood pressure
Patient cohort
Eligible patients were men or women ≥ 40 years of age with a diagnosis of PD, MSA, or PAF according to established consensus criteria [1, 8, 22]. Patients were mostly recruited from those followed in specialist autonomic clinics. Non-neurogenic causes for OH, including cardiac insufficiency or overuse of anti-hypertensive drugs, were ruled out following standard-of-care clinical guidelines. The diagnosis of nOH due to efferent autonomic failure was confirmed as appropriate on autonomic testing by (1) a fall in systolic BP > 30 mmHg within 5 min of tilt, (2) lack of phase IV BP overshoot after release of the Valsalva strain, and (3) reduced respiratory sinus arrhythmia in response to deep paced breathing [2, 23]. Other short-acting pressor agents were discontinued for ≥ 5 half-lives before first study dosing on day 1. Fludrocortisone could be continued at ≤ 0.1 mg/day. Long-acting antihypertensives were not allowed. Existing treatment with short-acting antihypertensives at bedtime for supine hypertension was permitted at the discretion of the investigator. Key exclusion criteria included systemic illnesses known to produce autonomic neuropathies such as diabetes, amyloidosis, and autoimmune conditions; known intolerance to NRIs; and use of any monoamine oxidase inhibitor within 14 days of first study dosing on day 1.
Assessments
Figure 1 shows the timing of all in- and outpatient assessments. BP was measured in the nondominant arm at heart level with an automated (or manual) sphygmomanometer after 5 and 10 min semi-supine (elevated 30°); after 5 and 10 min seated; and after 1, 3, 5, and 10 min standing immobile. In the dose-escalation trial, measurements were obtained predose and at 4, 7, 9, and 12 h postdose. In the double-blind treatment phase, measurements were obtained throughout the day. In the open-label extension phase, BP measurements were performed predose and at 4 h postdose.
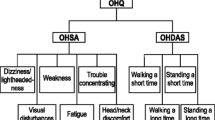
Symptom burden was evaluated using the validated Orthostatic Hypotension Questionnaire (OHQ) [24]. This included the six-item OH symptom assessment (OHSA) to capture the severity of cardinal symptoms of nOH on standing (e.g., dizziness/lightheadedness/near fainting, visual problems, weakness, fatigue, trouble concentrating, and head/neck discomfort) and the four-item Orthostatic Hypotension Daily Activities Scale (OHDAS) to capture interference of low BP symptoms on activities that require standing or walking. Participants were required to rate their symptoms on a Likert scale from 0 to 10, with 10 being most severe. Additional exploratory efficacy measures in the open-label extension phase included Patient Global Impression of Severity (PGI-S) [25] assessed on an 8-point Likert scale (from 0 = not assessed to 7 = very much worse).
Safety monitoring included AE reporting [including serious AEs (SAEs)], monitoring of vital signs (including supine BP), 12-lead electrocardiogram, clinical laboratory tests (hematology, serum chemistry, and urinalysis), use of concomitant medications, and physical examination.
Endpoints
The primary pharmacodynamic endpoint in the dose-escalation (Part A) and double-blind, placebo-controlled (Part B) inpatient studies was the change in seated systolic BP 6–8 h after drug administration. The primary pharmacodynamic endpoint in the open-label extension phase was an improvement from baseline in dizziness/lightheadedness scores (OHSA item 1) at the end of week 4 [24]. Other exploratory secondary endpoints included (1) change in the overall OHSA score, (2) change in the overall OHDAS score, (3) a change in the combined composite OHQ score, (4) change in standing systolic BP, (5) change in PGI-S, (6) duration of standing time, and (7) area under the curve for seated systolic BP 0–12 h after study drug. The pharmacokinetics and pharmacodynamics findings are reported elsewhere [21].
Statistical analysis
Planned enrollment in Part A was 40 participants. Planned enrollment in Part B was 20 participants (10 in the ampreloxetine treatment group and 10 in the placebo group). Planned enrollment for Part C was 20 participants. If 20 participants completed week 4, 13 responders (increase in OHSA item 1 score > 2) were needed to reject the null hypothesis of response rate < 40% (power 60.1%).
BP responders were defined as those with a ≥ 10 mmHg increase in seated systolic BP after ampreloxetine administration. Participants with OHSA item 1 score of > 4 at baseline were defined as symptomatic. Improvement of 1 point (− 1) on OHSA item 1 was applied as the minimal clinically important difference based on efficacy data from the literature (MCID) [11]. Composite OHQ scores were calculated as the sum of item scores divided by number of items, as described elsewhere [24]. PGI-S reported by participants was grouped into “improvement” or “no change/worsening” in the severity of nOH symptoms compared to baseline and summarized as participant counts and percentages [26]. In the dose-escalation study (Part A), paired t tests were used to compare time-matched differences between each ampreloxetine dose and placebo (day 1). In the 1 day, double-blind, placebo-controlled study (Part B), the difference in seated and standing systolic BP between ampreloxetine and placebo at 4–8 h after study drug administration was determined using a mixed model for repeated measures, with treatment group, time point, and interaction between the treatment group and time point as fixed factors. In the open-label extension phase (Part C), baselines for BP, OHSA, OHDAS, and PGI-S were determined after readmission before the first dose of ampreloxetine. All analyses in Part C were descriptive. No imputation for missing data was performed. Safety data for all studies were listed by participant and summarized using participant count and percentage. Participants with severe symptoms (defined as OHSA item 1 score > 4 points at baseline) were identified as a subgroup for additional analyses.
There was no formal hypothesis testing, and all results including P values were descriptive. Blood pressure data during Part B were tested for significance using Student’s t test, with the empirical assumption that the data were not normally distributed given the small number of subjects. Analysis was performed with SAS version 9.3 or later. Descriptive statistics by treatment are reported as mean ± SD; comparisons with placebo are reported as mean ± SEM unless otherwise stated.
Results
Patient characteristics and study flow
A total of 34 patients were enrolled, and 33 received at least one dose of ampreloxetine. The demographics are listed in Table 1 and reflected enrollment at specialized autonomic centers [23]. There was a predominance of white men. Mean age was 66 (range 51–83) years. Approximately 50% of participants met diagnostic criteria for MSA. Twenty-six out of 34 participants were unable to remain standing for 10 min (Table 1). The orthostatic heart rate rise was significantly impaired and fell within the diagnostic range for nOH [27]. Twenty-one participants were enrolled in the open-label extension phase (Part C), with 17 completing assessments at week 4, 12 completing week 20, and 11 completing week 24. Seventeen participants entering the open-label extension phase were determined to be highly symptomatic at baseline with an OHSA item 1 score > 4; of these, 13 completed week 4, 8 completed week 20, and 7 completed week 24.
Reasons for dropouts in the dose-escalation study (Part A) were physician decision (n = 3), an AE (n = 1), and withdrawal of consent (n = 1). No participant withdrew in the double-blind, placebo-controlled study (Part B). In the open-label extension phase (Part C), seven discontinuations were in participants with MSA, three of whom discontinued owing to physician decision, which most likely reflects the rapid progression of this disorder, and two discontinuations were owing to unrelated AEs and two were withdrawal by participant. Three non-MSA participants discontinued the study owing to physician decision. Five participants temporarily interrupted and then restarted ampreloxetine in Part C. Figure 2 describes the participant flow and reasons for dropouts.

Consort diagram describing the flow of participants through the study. *Participants who completed Part A and entered Part B before amendment. **Number of participants who entered Part C after completion of Part B (before amendment) and those who directly entered Part C from Part A (after amendment). mg milligrams, n number
Prior and concomitant medications
At enrollment (Part A), 24/34 (71%) participants had received any prior pressor medication. The most common prior pressor medications were midodrine (n = 17) and droxidopa (n = 8), which were stopped prior to start of study dosing. During the study, 17/34 (50%) received concomitant fludrocortisone. Three patients with MSA were receiving pharmacological treatment for supine hypertension (2 patients were treated with a 0.1 mg/h nitroglycerine transdermal patch at bedtime and 1 patient was treated with losartan).
Dose-escalation study (Part A)
Seventeen participants received 1 mg, 31 participants 2.5 mg, 29 participants 5 mg, 28 participants 10 mg, and 13 participants 20 mg (Part A). Figure 3a shows the increase in seated systolic BP relative to placebo at each dose. The percentage of responders (defined as an increase in seated systolic BP of ≥ 10 mmHg relative to placebo) at 6–8 h after study drug administration was highest at the 5 mg (43%) and 10 mg (39%) ampreloxetine doses. Responders did not escalate to higher doses. Of the 13 participants receiving the 20 mg maximal dose, no additional benefit to seated systolic BP was observed (Fig. 3a).

Pressor response to ampreloxetine. a Mean ± SE placebo time-matched change in seated SBP (primary endpoint) throughout dose escalation in the overall cohort (light blue) and responders (dark blue). Owing to an early trial amendment, not all participants received the 1 mg dose on day 1. Responders were not escalated to a higher dose. b Mean ± SE change in seated SBP 4 h after ampreloxetine (15 mg, n = 4, 10 mg, n = 1) vs. placebo (n = 5) in the 1 day double-blind, placebo-controlled randomized study. c Timeline of change in seated BP over 12 h after ampreloxetine and placebo. d Change in standing SBP 4 h after ampreloxetine and placebo. e Timeline of changes in standing SBP when assigned to ampreloxetine vs. placebo. Red dotted line denotes cutoff for a pressor responder, determined by a change in seated SBP ≥ 10 mmHg compared to baseline. In panels C through E, data shown as least squares mean ± SE; blue represents ampreloxetine, gray represents placebo. Red star indicates significance. BP blood pressure, mmHg millimeters of mercury, SBP systolic blood pressure, SE standard error
Double-blind, placebo-controlled study (Part B)
Ten participants were enrolled in the double-blind, placebo-controlled study (Part B). In the ampreloxetine treatment group, the most common dose was 15 mg (4 participants received 15 mg, 1 participant received 10 mg). As shown in Fig. 3b, 4 h after ampreloxetine, seated systolic BP increased by 15.7 mmHg compared to a decrease of 14.2 mmHg after placebo, yielding a least square mean difference of 29.9 mmHg (95% CI 7.6–52.3; P = 0.0112). An increase in standing BP was also observed with ampreloxetine (Fig. 3d). Four hours after ampreloxetine, standing systolic BP at minute 3 was numerically higher (35.0 ± 20.6 mmHg) than placebo (95% CI − 18.8 to 88.8). The pressor response subsided after 8 h and within 12 h was not different from placebo (Fig. 3c and e). In total, 4/5 of the patients receiving ampreloxetine reported a ≥ 1-point improvement in their OHSA item 1 score vs. 2/5 of the participants receiving placebo.

Symptoms and BP response over 20 weeks of open-label extension. a Mean change from baseline in dizziness/lightheadedness scores (OHQ symptom assessment item 1) in the subset of symptomatic participants (OHSA#1 > 4 points). A decrease in scores indicates clinical improvement. The primary endpoint was the change observed at week 4. Values below dotted red line indicate improvement above the MCID. The observed effect was sustained throughout the 20 weeks. After withdrawal of ampreloxetine, symptoms diminished to baseline levels. b Similar pattern of changes observed with the composite scores of symptom severity (OHSA) and impact of symptoms on activities of daily living (OHDAS). c Change from baseline in systolic BP at 1 min standing over the 20-week trial. Note, after withdrawal of ampreloxetine, most patients (N = 7) resumed taking other pressor agents. An increase in BP was observed off-treatment without corresponding improvement in symptoms. Data are mean ± standard error. BP blood pressure, MCID minimal clinically important difference, OHDAS Orthostatic Hypotension Daily Activities Scale, OHQ Orthostatic Hypotension Questionnaire, OHSA Orthostatic Hypotension symptom assessment, OHSA #1 dizziness/lightheadedness score on Orthostatic Hypotension Symptom Assessment (question 1)
Symptom improvement on open-label extension
A total of 21 patients were enrolled in the open-label extension (Part C). The median dose of ampreloxetine in the 20-week treatment phase was 10 mg. As shown in Fig. 4a, during the entire treatment duration of 20 weeks, average improvement in OHSA item 1 in the symptomatic subset was consistently > 1 point (MCID). Symptoms of dizziness/lightheadedness improved as early as week 1. Analysis of symptomatic patients (OHSA item 1 score > 4 at baseline) revealed a 3.8 ± 3.1 point (mean ± SD) decrease in dizziness/lightheadedness scores at the end of 4 weeks of treatment. On treatment, 77% of symptomatic participants reported ≥ 2-point improvement, 69% reported ≥ 3-point improvement, and 54% reported ≥ 4-point improvement at week 4. Symptomatic improvement was sustained at the end of week 20 with a mean decrease in symptom scores of − 3.1 ± 3.0 points (mean ± SD); 86% reported ≥ 1-point improvement, 71% reported ≥ 2-point improvement, and 43% of patients reported ≥ 4-point improvement. After ampreloxetine withdrawal, symptoms worsened and returned to pretreatment levels despite participants restarting alternative pressor agents. After 4 weeks of ampreloxetine withdrawal, mean (± SD) improvement had dropped to − 0.3 ± 1.9 points, and the proportion of participants reporting ≥ 1-point improvement dropped to 50%; no patient reported improvement of > 2 points. As expected, the nonsymptomatic subset also reported improvement, but the improvement was less pronounced than that seen with the symptomatic subset of participants.
Similarly, improvements in the OHSA and OHDAS composite scores were seen as early as week 1, were sustained throughout the 20 week treatment period, and diminished to pretreatment levels after ampreloxetine withdrawal (Fig. 4b).
Durability of the pressor response on open-label extension
Throughout the 20 weeks of treatment with ampreloxetine, standing systolic BP was increased from baseline. As shown in Fig. 4c, the pressor response was similar at week 4 (9.0 ± 23.6 mmHg) and week 20 (10.8 ± 12.1 mmHg). In the withdrawal phase, 7/11 (64%) patients resumed treatment with their other pressor agents (midodrine and droxidopa). Ampreloxetine withdrawal reversed the symptomatic benefit, with dizziness/lightheadedness scores (OHSA item 1) returning to baseline over the 4 weeks (Fig. 4a). The improvement in activities of daily living was lost after withdrawing ampreloxetine, despite most participants restarting other pressor agents (Fig. 4b). In the subset of participants who were symptomatic at baseline, average predose standing time was 5.3 ± 4.1 min (n = 17). In the open-label study, regression analysis of the pressor effect supine vs. standing showed a greater effect on BP in the upright position at week 4 and week 20 (Fig. 5a and b). At the end of the 20-week, open-label extension phase, standing time increased by 4 min, and participants were able to remain standing for an average of 8.6 ± 5.9 min (n = 7). After ampreloxetine withdrawal, the improvement in standing time was lost, and standing duration returned to baseline levels (4.8 ± 4.4 min, n = 6). In the 4 week withdrawal phase, only two participants were able to stand for 3 min to complete the BP measurement. At the end of the 4 weeks of withdrawal, symptoms had worsened ≥ 5 points in 80% of the participants who answered the global impression assessment scale (n = 8/10).

Responses in autonomic failure. a Change in SBP supine vs. change in 3-min standing SBP at week 4. Data are mean ± SD. A 5-mmHg increase in supine SBP was accompanied by an approximately 10-mmHg increase in standing SBP. b The durability of the pressor effect supine vs. standing was demonstrated to a greater degree on 3-min standing SBP at week 20. The increase in standing SBP exceeded the increase in supine SBP in participants receiving ampreloxetine. Red indicates before breakfast; blue indicates before lunch. Data from Part C; open-label extension. mmHg millimeters of mercury, pg picogram, SBP systolic blood pressure, SD standard deviation
Safety profile
Table 2 provides a full listing of AEs. Ampreloxetine was generally well tolerated. In the dose-escalation study (Part A), there were no SAEs. The most common AEs were headache (5.9%), urinary tract infection (2.9%), and constipation (5.9%); only 1 AE led to study drug discontinuation. In Part B, the only AE reported in a patient randomized to ampreloxetine was a urinary tract infection in a participant with MSA, which was considered to be unrelated to study drug. In the open-label extension phase (Part C), 18 of 21 participants reported at least 1 AE; the majority (61.9%) were moderate or severe. In Part C, five patients (23.8%) experienced at least one SAE, and none of the SAEs were considered related to study drug. No SAEs were of a psychiatric or cognitive nature. AEs led to permanent study discontinuation for two (9.5%) participants in Part C. The most common AEs were urinary tract infection (23.8%), hypertension (19.0%), and headache (14.3%) and were considered not related to ampreloxetine. No deaths were reported in this study. Overall, the most TEAEs considered to be study treatment related were headache in four participants (2 each in Part A and Part C) and hypertension in two participants (Part C).
Discussion
A pressor effect was observed with ampreloxetine treatment in the dose-titration phase, as evidenced by approximately 40% of participants experiencing a ≥ 10 mmHg increase in seated systolic BP relative to placebo. Moreover, in the double-blind study, BP was significantly higher on ampreloxetine compared to placebo. There was durable improvement in symptoms over 20 weeks of open-label treatment, which worsened back to baseline levels once ampreloxetine was withdrawn.
These findings are encouraging. To date, there are no available therapies for nOH in patients with autonomic synucleinopathies that have shown a durable effect beyond 1 week in clinical trials. Therapies with approved available pressor agents (midodrine and droxidopa) have been shown to exacerbate supine hypertension [12, 15], which is a limiting factor in achieving therapeutic benefit [13, 28]. The mechanism of action of ampreloxetine as an NRI provides an alternative strategy to available pressor agents [17]. Plasma measurements of NE in this target population suggest that ampreloxetine blocks NE reuptake upon release, increasing its bioavailability at the neurovascular junction, and thus may harness the effect of the residual postganglionic sympathetic neurons preferentially when activated by the baroreflex on standing [21]. This is a more physiological approach that may reduce the risk of supine hypertension, as it would be most likely to potentiate sympathetic tone when activated upon standing [2]. The selectivity of ampreloxetine for the NET over the serotonin transporter and its long plasma half-life with stable plasma levels over 24 h suggest that it may be of potential benefit as a once-daily, durable treatment for nOH [20, 29]. We did not formally restrict recruitment to patients who failed treatment with other pressor agents as we wanted a representative sample of typical patients with nOH at various stages of disease.
Ampreloxetine had a favorable safety profile in participants with nOH. The most common AEs were urinary tract infections, which occur frequently in patients with autonomic synucleinopathies owing to bladder dysfunction, especially those with MSA, who made up the largest proportion of patients recruited into the trial [30]. Supine hypertension, particularly at night, is inherent to this group of disorders with or without pressor agent therapy [13, 28]. Although no signal was observed that ampreloxetine worsened supine hypertension at 4 and 20 weeks of treatment (and preferentially impacted standing BP; Fig. 5), hypertension was a reported AE in four patients (19%), but only in Part C, and this effect of ampreloxetine will continue to be investigated in future studies. Of note, data were only available for seven participants at week 20; longer-term studies with larger numbers of participants are ongoing to continue to investigate.
Ampreloxetine doses between 5 and 10 mg had the highest percentage of pressor responders relative to placebo (Fig. 3). A further increase in the proportion of BP responders was not observed at the 20 mg dose and may be due to steady-state concentrations being reached as preliminary pharmacokinetic analysis of participants in Part C showed that steady-state concentrations were achieved between days 1 and 15. These results, combined with the lack of any appreciable difference in safety or tolerability between 5 and 10 mg doses, led to the decision to proceed at a dose of 10 mg in the phase 3 studies. The pressor response to ampreloxetine and improvement in symptoms in the double-blind, placebo-controlled study (Part B), based on ten participants, provides an encouraging signal for efficacy.
The results of the open-label extension phase (Part C), based on 17 participants who were symptomatic at baseline, suggest that symptomatic benefit is achieved within 1 week and is durable for up to 20 weeks of treatment. In the withdrawal phase, despite restarting other pressor agents, symptoms worsened back to baseline after ampreloxetine was discontinued, which suggests that the symptom-related benefit may have been specific to ampreloxetine. We did not see a clear decrease in blood pressure after ampreloxetine withdrawal, probably owing to most patients (n = 7) restarting their previous medications for nOH. Two previous studies have shown that although midodrine and NET inhibitors both improved orthostatic BP, NET inhibition was better in terms of improving symptoms in participants with nOH [19, 31].
We did not detect a difference in clinical benefit between participants with a central preganglionic phenotype of autonomic failure (e.g., MSA), who are presumed to have sparing of postganglionic sympathetic fibers, and those with Lewy body disorders, in which the brunt of the α-synuclein deposition is thought to occur in the postganglionic fibers (i.e., PD and PAF) [7, 17, 23, 30]. This may be because the sample size was too small to detect a clinical effect by disease state. It may also be that the neuropathological phenotypes of autonomic failure are less precise or frequently mixed according to disease state. Many patients with PD and PAF have some sparing of the postganglionic sympathetic nerve fibers combined with denervation super-sensitivity within the vasculature [10]. As is observed with other pressor agents [10, 32], the levels of NE and the degree of residual sympathetic vasoconstrictor tone may determine the response to treatment rather than the diagnosis per se. In line with this assumption, we found that the pressor response to standing following ampreloxetine was related to the levels of NE (Fig. 5), which is a biomarker of overall residual sympathetic tone in patients with autonomic failure [2, 32, 33]. We will continue to explore the hypothesis that the extent of denervation drives the clinical responsiveness to NET inhibition in our larger patient cohort.
Our study has some limitations, including the small number of participants, particularly in Part B. Furthermore, the 20-week treatment (Part C) was an open-label extension with no placebo control. The prolongation of standing time after treatment with ampreloxetine and worsening after withdrawal is an encouraging signal; however, a larger cohort is needed to confirm this. Enrollment was limited to specialist autonomic clinics because we wanted to carefully observe the findings. The fall in BP 4 h after placebo observed in Part B (Fig. 3c and e) could have had a number of causes including small sample size, and because of variation in the timing of meals, it was not possible to determine whether it was caused by postprandial splanchnic vasodilatation. A larger sample size is needed to better understand whether the AEs related to hypertension are de novo, exacerbation of preexisting supine hypertension, or part of normal disease variability. At week 12 (Fig. 4c), there was not always a tight correlation between the degree of hypotension and severity of orthostatic symptoms, which could be due to several physiologic factors including cerebral autoregulation. The ampreloxetine phase 3 program includes and should allow us to examine factors that influence responsiveness to ampreloxetine in a larger sample size of patients. The dropouts seen throughout the trial likely reflect the high proportion of MSA participants enrolled in the phase 2 program and the rapid worsening of the participants’ underlying neurological disease and progressive difficulty with mobility and attending in-clinic follow-up visits. This underscores the need for a decentralized trial design as a retention strategy with remote visits for future clinical trials.
In conclusion, the results of this small phase 2 clinical trial suggest that once-daily 10 mg ampreloxetine is generally safe and well tolerated and may have a durable clinical benefit over 5 months of treatment. The data from this study informed the design and initiation of two placebo-controlled phase 3 clinical trials and an extension study to further evaluate the longer-term efficacy and safety of a once-daily 10 mg dose of ampreloxetine in patients with symptomatic nOH (NCT03750552, NCT03829657, and NCT04095793). The phase 3 program includes and should allow us to examine factors that influence responsiveness to ampreloxetine in a larger sample size of patients.
Data availability
Theravance Biopharma (and its affiliates) will not be sharing individual deidentified participant data or other relevant study documents.
References
Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, Cheshire WP, Chelimsky T, Cortelli P, Gibbons CH, Goldstein DS, Hainsworth R, Hilz MJ, Jacob G, Kaufmann H, Jordan J, Lipsitz LA, Levine BD, Low PA, Mathias C, Raj SR, Robertson D, Sandroni P, Schatz I, Schondorff R, Stewart JM, van Dijk JG (2011) Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res 21:69–72. https://doi.org/10.1007/s10286-011-0119-5
Kaufmann H, Norcliffe-Kaufmann L, Palma JA (2020) Baroreflex dysfunction. N Engl J Med 382:163–178. https://doi.org/10.1056/NEJMra1509723
Low PA (2008) Prevalence of orthostatic hypotension. Clin Auton Res 18(Suppl 1):8–13. https://doi.org/10.1007/s10286-007-1001-3
Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, Martinez J, Tijero B, Berganzo K, Kaufmann H (2015) Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Mov Disord 30:639–645. https://doi.org/10.1002/mds.26079
Ganz DA, Latham NK (2020) Prevention of falls in community-dwelling older adults. N Engl J Med 382:734–743. https://doi.org/10.1056/NEJMcp1903252
Claassen DO, Adler CH, Hewitt LA, Gibbons C (2018) Characterization of the symptoms of neurogenic orthostatic hypotension and their impact from a survey of patients and caregivers. BMC Neurol 18:125. https://doi.org/10.1186/s12883-018-1129-x
Hague K, Lento P, Morgello S, Caro S, Kaufmann H (1997) The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol 94:192–196. https://doi.org/10.1007/s004010050693
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H (2008) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 131:642–650. https://doi.org/10.1093/brain/awm302
Shibao CA, Biaggioni I (2020) Management of orthostatic hypotension, postprandial hypotension, and supine hypertension. Semin Neurol 40:515–522. https://doi.org/10.1055/s-0040-1713886
Biaggioni I, Arthur Hewitt L, Rowse GJ, Kaufmann H (2017) Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol 17:90. https://doi.org/10.1186/s12883-017-0867-5
Wright RA, Kaufmann HC, Perera R, Opfer-Gehrking TL, McElligott MA, Sheng KN, Low PA (1998) A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology 51:120–124. https://doi.org/10.1212/wnl.51.1.120
Jordan J, Fanciulli A, Tank J, Calandra-Buonaura G, Cheshire WP, Cortelli P, Eschlboeck S, Grassi G, Hilz MJ, Kaufmann H, Lahrmann H, Mancia G, Mayer G, Norcliffe-Kaufmann L, Pavy-Le Traon A, Raj SR, Robertson D, Rocha I, Reuter H, Struhal W, Thijs RD, Tsioufis KP, Gert van Dijk J, Wenning GK, Biaggioni I (2019) Management of supine hypertension in patients with neurogenic orthostatic hypotension: scientific statement of the American Autonomic Society, European Federation of Autonomic Societies, and the European Society of Hypertension. J Hypertens 37:1541–1546. https://doi.org/10.1097/HJH.0000000000002078
Grijalva CG, Biaggioni I, Griffin MR, Shibao CA (2017) Fludrocortisone is associated with a higher risk of all-cause hospitalizations compared with midodrine in patients with orthostatic hypotension. J Am Heart Assoc 6:e006848. https://doi.org/10.1161/JAHA.117.006848
McDonell KE, Preheim BA, Diedrich A, Muldowney JAS 3rd, Peltier AC, Robertson D, Biaggioni I, Shibao CA (2019) Initiation of droxidopa during hospital admission for management of refractory neurogenic orthostatic hypotension in severely ill patients. J Clin Hypertens (Greenwich) 21:1308–1314. https://doi.org/10.1111/jch.13619
Shibao C, Raj SR, Gamboa A, Diedrich A, Choi L, Black BK, Robertson D, Biaggioni I (2007) Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension 50:47–53. https://doi.org/10.1161/HYPERTENSIONAHA.107.089961
Jordan J, Shibao C, Biaggioni I (2015) Multiple system atrophy: using clinical pharmacology to reveal pathophysiology. Clin Auton Res 25:53–59. https://doi.org/10.1007/s10286-015-0271-4
Schroeder C, Jordan J (2012) Norepinephrine transporter function and human cardiovascular disease. Am J Physiol Heart Circ Physiol 303:H1273-1282. https://doi.org/10.1152/ajpheart.00492.2012
Ramirez CE, Okamoto LE, Arnold AC, Gamboa A, Diedrich A, Choi L, Raj SR, Robertson D, Biaggioni I, Shibao CA (2014) Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension 64:1235–1240. https://doi.org/10.1161/HYPERTENSIONAHA.114.04225
Kanodia J, Lo A, Baldwin RM, Graham RA, Bourdet DL (2021) Pharmacokinetics of ampreloxetine, a norepinephrine reuptake inhibitor, in healthy subjects and adults with attention-deficit/hyperactive disorder or fibromyalgia pain. Clin Pharmacokinet 60:121–131. https://doi.org/10.1007/s40262-020-00918-7
Lo A, Norcliffe-Kaufmann L, Vickery R, Bourdet D, Kanodia J (2021) Pharmacokinetics and pharmacodynamics of ampreloxetine, a novel, selective norepinephrine reuptake inhibitor, in symptomatic neurogenic orthostatic hypotension. Clin Auton Res 31:395–403. https://doi.org/10.1007/s10286-021-00800-x
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184. https://doi.org/10.1136/jnnp.55.3.181
Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, Goldstein DS, Peltier AC, Shibao CA, Gibbons CH, Freeman R, Robertson D, Autonomic Disorders Consortium (2017) Natural history of pure autonomic failure: a United States prospective cohort. Ann Neurol 81:287–297. https://doi.org/10.1002/ana.24877
Kaufmann H, Malamut R, Norcliffe-Kaufmann L, Rosa K, Freeman R (2012) The orthostatic hypotension questionnaire (OHQ): validation of a novel symptom assessment scale. Clin Auton Res 22:79–90. https://doi.org/10.1007/s10286-011-0146-2
Busner J, Targum SD (2007) The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont) 4:28–37
Guy W (1976) ECDEU assessment manual for psychopharmacology, revised. Rockville, MD: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs
Norcliffe-Kaufmann L, Kaufmann H, Palma JA, Shibao CA, Biaggioni I, Peltier AC, Singer W, Low PA, Goldstein DS, Gibbons CH, Freeman R, Robertson D, Autonomic Disorders Consortium (2018) Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies. Ann Neurol 83:522–531. https://doi.org/10.1002/ana.25170
Palma JA, Redel-Traub G, Porciuncula A, Samaniego-Toro D, Millar Vernetti P, Lui YW, Norcliffe-Kaufmann L, Kaufmann H (2020) The impact of supine hypertension on target organ damage and survival in patients with synucleinopathies and neurogenic orthostatic hypotension. Parkinsonism Relat Disord 75:97–104. https://doi.org/10.1016/j.parkreldis.2020.04.011
Smith JA, Bourdet DL, Daniels OT, Ding YS, Gallezot JD, Henry S, Kim KH, Kshirsagar S, Martin WJ, Obedencio GP, Stangeland E, Tsuruda PR, Williams W, Carson RE, Patil ST (2014) Preclinical to clinical translation of CNS transporter occupancy of TD-9855, a novel norepinephrine and serotonin reuptake inhibitor. Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyu027
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372:249–263. https://doi.org/10.1056/NEJMra1311488
Byun JI, Kim DY, Moon J, Shin HR, Sunwoo JS, Lee WJ, Lee HS, Park KI, Lee ST, Jung KH, Jung KY, Kim M, Lee SK, Chu K (2020) Efficacy of atomoxetine versus midodrine for neurogenic orthostatic hypotension. Ann Clin Transl Neurol 7:112–120. https://doi.org/10.1002/acn3.50968
Palma JA, Norcliffe-Kaufmann L, Martinez J, Kaufmann H (2018) Supine plasma NE predicts the pressor response to droxidopa in neurogenic orthostatic hypotension. Neurology 91:e1539–e1544. https://doi.org/10.1212/WNL.0000000000006369
Norcliffe-Kaufmann L, Katz SD, Axelrod F, Kaufmann H (2015) Norepinephrine deficiency with normal blood pressure control in congenital insensitivity to pain with anhidrosis. Ann Neurol 77:743–752. https://doi.org/10.1002/ana.24377
Acknowledgements
The authors thank the following people for their diligence and comprehensive oversight of the study; their contributions and suggestions helped lead to a successful study outcome: Jose-Alberto Palma, MD; Meredith Bryarly, MD; Jose Martinez, MA; Bonnie Black, RN; and Steve Hopkins, BS, CCRC. The authors thank Susan Manganaro, MD, MBA, Theravance Biopharma US, Inc., for assistance with data analysis and interpretation and manuscript revision and Kirsty Nahm, MD, formerly of The Curry Rockefeller Group, LLC (Tarrytown, NY), who provided medical writing and editorial assistance for the first draft, which was funded by Theravance Biopharma US, Inc.
Funding
This study was funded by Theravance Biopharma R&D, Inc.
Author information
Authors and Affiliations
Contributions
All authors were involved in the analysis and interpretation of these data, participated in the drafting of the manuscript, and have approved the final draft for journal submission.
Corresponding author
Ethics declarations
Conflict of interest
HK was a consultant to and has received research support from Theravance Biopharma US, Inc. RV is an employee of Theravance Biopharma Ireland Limited and stockholder of Theravance Biopharma, Inc. WW is a former employee of Theravance Biopharma US, Inc. JK is an employee of Theravance Biopharma US, Inc. and a stockholder of Theravance Biopharma, Inc. CS has received research support from Theravance Biopharma and is a member of the Advisory Board of Lundbeck US. LNK is an employee of Theravance Biopharma US, Inc. BH is a former employee of Theravance Biopharma UK Limited and a stockholder of Theravance Biopharma, Inc. IB is a consultant for and has received research support from Theravance Biopharma and is a patent holder of an automated abdominal binder for the treatment of orthostatic hypotension.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Local IRB approvals were obtained.
Consent to participate
Informed consent was obtained from all individual participants in the study.
Consent for publication
All authors have approved the final draft and agree to have these data published.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaufmann, H., Vickery, R., Wang, W. et al. Safety and efficacy of ampreloxetine in symptomatic neurogenic orthostatic hypotension: a phase 2 trial. Clin Auton Res 31, 699–711 (2021). https://doi.org/10.1007/s10286-021-00827-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10286-021-00827-0