
Abstract
Cell-to-cell transport of molecules in plants must be properly regulated for plant growth and development. One specialized mechanism that plants have evolved involves transport through plasmodesmata (PD), but when and how transport of molecules via PD is regulated among individual cells remains largely unknown, particularly at the single-cell level. Here, we developed a tool for quantitatively analyzing cell-to-cell transport via PD at a single-cell level using protonemata of Physcomitrella patens and a photoconvertible fluorescent protein, Dendra2. In the filamentous protonemal tissues, one-dimensional intercellular communication can be observed easily. Using this system, we found that Dendra2 was directionally transported toward the apex of the growing protonemata. However, this directional transport could be eliminated by incubation in the dark or treatment with a metabolic inhibitor. Thus, we propose that directional transport of macromolecules can occur via PD in moss protonemata, and may be affected by the photosynthetic and metabolic activity of cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intercellular communication is essential for coordinated growth, development, defense, and responses to the environment in multi-cellular organisms. In plants, neighboring cells are connected directly via cell wall spanning, plasma membrane-lined channels called plasmodesmata (PD) (Burch-Smith and Zambryski 2012; Maule 2008). PD enable intercellular movement of micromolecules and macromolecules, such as water, ions, metabolites, proteins, RNAs and plant viruses, and this molecular movement is tightly regulated in growth, development and the response to environmental stresses (Burch-Smith et al. 2011; Maule 2008; Zambryski and Crawford 2000).
Intercellular movement of molecules via PD can be controlled by the PD size exclusion limit (SEL), defined as the size of the largest molecules that can diffuse through the PD. The PD SEL is an important regulator and can be spatiotemporally controlled throughout plant development, resulting in the establishment of symplastic fields (Burch-Smith et al. 2011; Roberts and Oparka 2003). In these fields, particular cells can be symplastically isolated from surrounding cells within tissues and organs (Roberts and Oparka 2003), and the isolated groups of cells can initiate independent developmental programs (Duckett et al. 1994; Gisel et al. 2002; Kim et al. 2005; Kobayashi et al. 2005). Furthermore, a single cell also can be symplastically isolated in specific developmental programs, e.g. in root hairs (Duckett et al. 1994), stomatal guard cells (Palevitz and Hepler 1985) and cotton fiber cells (Ruan et al. 2001). Thus, intercellular movement of molecules must be regulated not only at the tissue level but also at the cellular level to specify cell fates and patterning during plant development.
Previous studies have suggested that the manner of molecular movement through PD can be separated into two modes, targeted and non-targeted movement (Crawford and Zambryski 2001). In targeted movement, specific proteins such as transcriptional factors, including SHORT-ROOT and CAPRICE, and plant viral movement proteins actively increase the PD SEL to allow their passage into neighboring cells (Epel 2009; Wu and Gallagher 2011), and are directionally transported to their target cells. By contrast, in non-targeted movement, many micromolecules and macromolecules can simply diffuse through the PD and thus non-targeted movement is governed by endogenous PD SEL (Wu et al. 2003; Wu and Gallagher 2011).
Non-specific tracers such as exogenous fluorescent dyes and proteins have been used to dissect developmental, spatio-temporal and physiological regulation of non-targeted movement of molecules through PD (Crawford and Zambryski 1999; Kim and Zambryski 2005). For analysis of non-targeted movement via PD at a cellular level, several methods to introduce tracers into a single cell have been established (Oparka and Boevink 2005). In early studies, microinjection and particle bombardment were used extensively for analysis of non-targeted movement of molecules in various plant tissues (Dashevskaya et al. 2008; Ehlers and van Bel 2010; Liarzi and Epel 2005; Palevitz and Hepler 1985; Tucker 1982), but these methods are invasive and damaging to cells, and have been known to lead to the artificial modification of PD transport by cellular wounding (Oparka and Prior 1992). For a non-invasive method, cell-specific-promoters can be used for expression of fluorescent proteins within a specific single cell to analyze PD transport from that cell to its neighbors (Imlau et al. 1999; Oparka et al. 1999); however, the available cells for analysis are limited to the cells for which specific promoters have been indentified. Therefore, recently, photoactivatable tracers such as PA-GFP or caged fluorescein have been used for analysis of non-targeted movement of molecules via PD at the cellular level (Christensen et al. 2009; Liesche and Schulz 2012a; Martens et al. 2004). These tracers can be photoactivated by UV light to non-invasively label any single cell within a tissue, which enables us to observe PD transport from the labeled cell to its neighbors. Using caged fluorescein, Liesche and Schulz (2012b) developed a method to quantify PD-mediated cell-wall permeability at the cellular level in Cucurbita maxima leaves. It is, however, yet difficult to irradiate UV light into a single target cell and to accurately trace three-dimensional cell-to-cell movement of tracers in complex, three-dimensionally organized tissues.
For the analysis of non-targeted movement of molecules via PD at a single cell level, filamentous tissues such as multi-cellular trichomes (Christensen et al. 2009; Waigmann and Zambryski 2000) and stamen hairs (Radford and White 2011; Tucker 1982) can be powerful tools, because it is easy to observe the one-dimensional intercellular communication in the filament, and to introduce tracers into a single cell. With such advantages, Tucker et al. (1989) developed a method in which the kinetics of PD transport between individual cells could be measured using stamen hairs. Furthermore, by introduction of tracers into any single cell in trichomes of tobacco leaves to analyze PD transport between individual cells, unidirectional transport across the epidermal/trichome boundary was found, which appears to be important in the establishment of the symplastic fields of trichomes (Christensen et al. 2009; Waigmann and Zambryski 1995). Thus, simple filamentous tissues allow us to simplify the analysis of PD transport at the cellular level. Until now, however, reports have been confined to a few kinds of filamentous tissues such as stamen hairs and trichomes.
Here, we propose a novel tool for analyzing non-targeted movement of macromolecules via PD at the cellular level, by using the filamentous protonemal tissues of the model moss Physcomitrella patens (Cove et al. 2006). Protonemata are composed of single files of cells, which are particularly amenable for studies of cell growth and differentiation. Protonemata grow by division of the apical cells to produce a developmental gradient with younger cells at the apex and more differentiated cells toward the base (Cove et al. 2006; Duckett et al. 1998; Pressel et al. 2008). In moss protonemata, PD ultrastructure has been studied by observation with transmission electron microscopy, which indicated that abundant seed-plant-like PD exist within the septum between protonemal cells (Cook et al. 1997; Schnepf and Reinhard 1997; Schnepf and Sawidis 1991), although the branched PD observed in differentiated cells of angiosperms have not been found yet (Burch-Smith et al. 2011). However, little has been reported about intercellular movement of molecules via PD and its regulation in the moss (Rydin and Clymo 1989). To establish a tool to study cell-to-cell movement of molecules via PD at a cellular level, we visualized macromolecular movement between protonemal cells using the photoconvertible fluorescent protein, Dendra2 (Chudakov et al. 2007). We found that Dendra2 moved in the apical direction more readily than in the basal direction along protonemata. This directional transport was, however, eliminated by incubation in the dark or treatment with the metabolic inhibitor sodium azide.
Materials and methods
Plant materials and growth conditions
Protonemal cells of wild-type of Physcomitrella patens Bruch & Schimp subsp. patens (Ashton and Cove 1977) and the transformants expressing Dendra2 were cultivated aseptically on BCDATG agar medium under continuous white light at 25 °C (Nishiyama et al. 2000).
Plasmid construction for EF1α:Dendra2
The open reading frame of Dendra2 was PCR-amplified with the appropriate primers (5′-CACCATGAACACCCCGG-3′ and 5′-TTAAGCTTGAGCTCGAGTCTTGTAC-3′) from the pDendra2-C vector (Chudakov et al. 2007). The open reading frame of Dendra2 was cloned into the pENTR/D-TOPO vector (Invitrogen, Tokyo, Japan) to generate the plasmid pENTR Dendra2. The resultant plasmid pENTR Dendra2 was subjected to LR reaction using the destination vector pT1OG (Aoyama et al. 2012) to constitutively express Dendra2 under the control of the constitutive promoter (PpEF1-α promoter region; Scaffold_7: 2739557-2740907 of P. patens genome annotation v1.6; http://www.cosmoss.org/). The generated construct was digested with the restriction enzyme Sse8387I (Takara Bio, Ohtsu, Japan) for gene targeting and introduced into wild-type P. patens by polyethylene glycol (PEG)-mediated transformation as described previously (Nishiyama et al. 2000).
Photoconversion and time-lapse imaging
Photoconversion of Dendra2 in a protonemal cell was carried out using a laser scanning microscope, the Zeiss LSM510 META (Carl Zeiss, Tokyo, Japan). After selecting the target cell, photoconversions by 405 nm diode laser (0.3 % laser output, 50 mW) were done with 3 iterations (scanning speed 163.83 μs per pixel, zoom 12× and interval 10 s) using the plan-apochromat 10×/0.45 NA objective. The 10th or more basal protonemal cell, as counted from the apical cell, was mainly selected to be photoconverted, since Dendra2 mobility was too low to evaluate the directional movement in cells apical to the 10th cell.
For time-lapse imaging, protonemal cells were cultured on BCDATG gellan gum medium, which contains 0.5 % gellan gum (Wako, Osaka, Japan) instead of agar, in a 35 mm Petri dishes with a 27 mm coverslip window at the bottom (Matsunami Glass Industries, Kishiwada, Japan). Since the Dendra2 in the protonemal cells was spontaneously photoconverted under white fluorescent light, we used protonemal cells cultured for 7–10 days under red light filtered through a red acrylic filter (Shinkolite A, no. 102, 3 mm thick, Mitsubishi Rayon, Tokyo, Japan) to avoid exposure to shorter wavelengths from white fluorescent light. We used a 561 nm diode-pumped solid-state (DPSS) laser at 15 mW (5 % power) for time-lapse imaging. The range of wavelengths used to capture the emission of red fluorescence of Dendra2 is 575–615 nm. Images were acquired at a resolution of 1,024 × 1,024 pixels with a scan rate of 0.8 μs per pixel using a plan-apochromat 10×/0.45 NA objective. Manual focusing was used to control the image plane. The time-lapse imaging was conducted at intervals of 15 min for 1 h.
For investigation of the effect of dark culture condition on Dendra2 movement in protonemata, a petri dish in which protonemata were cultured for 7–10 days under red light was wrapped in aluminum foil and incubated in a black-lacquered box for 24 h immediately before time-lapse imaging. For investigation of the effect of metabolic inhibitor on Dendra2 movement, protonemata were cultured for 7–10 days under red light and then sodium azide (30 μM, 0.1 mM, 1 mM and 10 mM, Nakalai Tesque, Kyoto, Japan, 1 M stock in water) was added to the medium surrounding protonemata 2 h before the start of time-lapse imaging.
The protonemata used for time-lapse imaging were subsequently cultured overnight (12–18 h) in the treatment conditions (red light, dark or NaN3) to observe the long-term movement of Dendra2. Dendra2 movement along these protonemata was imaged with a Leica DMLB (Leica, Tokyo, Japan) epifluorescence microscope equipped with a CoolSNAP Color digital camera (Nippon Roper, Tokyo, Japan). For detecting the red fluorescence of Dendra2, N3 filters (excitation 546/12 nm, emission 600/40 nm) were used.
Quantitative analysis of imaging data
The time-lapse images were analyzed using ImageJ 1.41o (Abramoff et al. 2004). The outlines of two cells adjacent to the photoconverted cell were traced manually. Then the mean fluorescence intensities per pixel were measured within the outlined areas for all images in the time sequence. The mean fluorescence intensities per pixel of areas where there was no protonemal cells were measured as the backgrounds in each image. The mean background intensities per pixel were subtracted from the mean fluorescence intensities per pixel of the outlined cells for each image to obtain the net values of the mean fluorescence intensities per pixel of the cells. Time-course of the net values of the mean fluorescence intensities per pixel of cells was plotted on a graph. A linear regression line (Excel, Microsoft, Tokyo, Japan) was fitted to each graph, and the slope of each regression line was calculated as the movement rate of Dendra2.
For analysis of long-term movement of Dendra2 along protonemata, captured images were analyzed using ImageJ. The outlines of a photoconverted cell and its four neighboring cells, which are Cell [+2], Cell [+1], Cell [−1] and Cell [−2] (see a frame format of protonemata in Fig. 4), were traced manually. Then the mean fluorescence intensities per pixel were measured within the outlined areas for all images. As with the analysis of time-lapse data, the net values of the mean fluorescence intensities per pixel of the cells were calculated by subtracting the mean background intensities per pixel from the mean fluorescence intensities per pixel of the outlined cells for each image.
Immunoblot analysis
Proteins were extracted from the 4-day-old protonemata in sample buffer containing 4 % (w/v) sodium dodecyl sulfate, 20 % (v/v) glycerol, 125 mM Tris–HCl pH 6.8, 0.2 M dithiothreitol and the complete mini protease inhibitor cocktail (Roche, Tokyo, Japan). The lysate was boiled for 5 min, and cleared of insoluble material by centrifugation. Extracted protein was separated by SDS-PAGE (Laemmli 1970) and transferred to Hybond-P membranes (GE Healthcare, Tokyo, Japan). After blocking with 5 % (w/v) skim milk (Meiji, Tokyo, Japan) in TTBS containing 20 mM Tris (pH 7.5), 150 mM NaCl, 0.1 % (v/v) Tween 20 at 4 °C overnight, the membrane was incubated with anti-Dendra2 (Evrogen, Moscow, Russia), diluted 1:5,000 with TTBS, for 1 h at room temperature. The membrane was then washed with TTBS, followed by incubation with ECL Rabbit IgG, HRP-Linked Whole antibody (GE Healthcare), diluted 1:5,000 with TTBS, for 1 h at room temperature. After washing with TTBS, signals from the secondary antibody were detected with ECL Plus Western Blotting Detection reagents (GE Healthcare).
Statistical analysis
Data of multiple groups were analyzed by one-way analysis of variance with a post hoc multiple comparison test (Tukey–Kramer procedure).
Results
Constitutive expression of Dendra2 for visualizing in vivo cell-to-cell transport
We set out to visualize the cell-to-cell movement of macromolecules via PD; to do this, we used time-lapse imaging in the protonemata of P. patens. As a tracer, we used the monomeric photoconvertible fluorescent protein Dendra2, which can be irreversibly photoconverted from a green to a red fluorescent state by excitation of the chromophore with blue light (optimum at 405 nm) (Chudakov et al. 2007). We generated transgenic strains expressing Dendra2 under a constitutive promoter and selected a transformant expressing Dendra2 at an appropriate level for imaging. The morphology and growth of the transformant was indistinguishable from wild type (Figs. 1a, S1). Dendra2 was expressed in whole protonemata and localized in the cytoplasm and the nuclei (Fig. 1b) and there was no red fluorescence before photoconversion of Dendra2 (Fig. 1c). Proper expression of Dendra2 in the protonemata was also confirmed by immunoblotting and the protein was found to be the expected size of 26 kD (Fig. S2). Dendra2 in a specific protonemal cell was photoconverted by illumination with 405 nm light (Fig. 1b, box) causing the cell to be labeled in red (Fig. 1f, g). Thus, we developed a transgenic plant where the fluorescent marker in a specific cell can be selectively photoconverted from green to red at will.

Photoconversion of Dendra2 expressed in a protonemal cell. Shown are images of the bright field (a, d), green fluorescence (b, e), red fluorescence (c, f) and overlay (g) for representative protonemal cells expressing Dendra2 (color figure online). b Dendra2 is localized in the cytoplasm and the nuclei of the protonemata. The box in (b) indicates the area where Dendra2 was photoconverted by irradiation at 405 nm with a diode laser (0.3 % laser power, 50 mW). f A cell (arrowhead) was selectively photoconverted from green to red. Scale bars 100 μm
Directional transport of macromolecules in growing protonemata
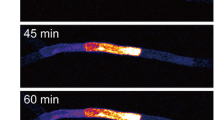
In order to observe the cell-to-cell movement of macromolecules in protonemata, we photoconverted Dendra2 in a selected protonemal cell (Fig. 2a, bright field image; Cell [0]) and then observed Dendra2 movement to the neighboring cells (apical side; Cell [+1], basal side; Cell [−1]) using time-lapse imaging (Fig. 2a, fluorescence images). We found that Dendra2 protein moved from the photoconverted cell to its neighboring cells. To quantitatively analyze the cell-to-cell movement of Dendra2, we measured and graphed the mean fluorescence intensities per pixel of Cell [+1] and Cell [−1] at each time point (Fig. 2b). The regression lines were plotted and their slopes were calculated to give the movement rate of Dendra2 in the apical and basal directions (V A and V B , respectively). In this case, V A (1.16 min−1) is larger than V B (0.57 min−1), indicating that Dendra2 moved faster toward the apical side.

Cell-to-cell movement of Dendra2 between protonemal cells. a A representative bright field image of the photoconverted cell (Cell [0]) and its neighboring cells (apical side; Cell [+1], basal side; Cell [−1]) in a protonema of the transformant expressing Dendra2, and a representative series of successive fluorescence images (0–60 min) showing the cell-to-cell movement of Dendra2 from the photoconverted cell to its neighboring cells. The protonemal apex is located to the left side. The time-lapse imaging was started within 30 min of photoconversion, and conducted for 60 min at intervals of 15 min. The starting timepoint of the time-lapse imaging is t = 0. In the time-lapse images, only red fluorescence is shown. Arrowheads represent the septum of protonemal cells. Scale bar 100 μm. b The kinetics of the mean fluorescence intensity per pixel of Cell [+1] and Cell [−1] are graphically shown (open circle and filled circle, respectively). The fluorescence intensity per pixel of each cell was measured with ImageJ. A regression line fitted to each set of data is shown, along with the equation and correlation coefficient (r 2). The slope of each regression line was calculated as the rate of Dendra2 movement in the apical and basal direction (V A and V B , respectively)
We next analyzed Dendra2 movement in many protonemata, and plotted the V B values as a function of V A values (Fig. 3a). We found that V A values tended to be larger than V B values (13 out of 16 showed V A > V B ), suggesting that Dendra2 can move faster in the apical direction than the basal direction in growing protonemata cultured under red light.

Relationship between V A and V B values depending on light conditions and metabolic activity. V A values are plotted as a function of V B values for protonemata cultured under red light (a), incubated in the dark for 24 h (b) or treated with 30 μM NaN3 for 2 h (c). A thin line is drawn in the panel passing through the origin, with a slope of 45°. Thus, V A values that are larger than V B values are located below this line. n = 16 (red light), 14 (dark) and 14 (NaN3)
Directional transport requires cellular photosynthetic and metabolic activity
To examine whether directional transport was affected by cell growth or cellular metabolic status, we analyzed Dendra2 movement in the protonemata after incubation in the dark for 24 h, where the protonemal growth was completely arrested (Fig. S3). We determined V A and V B values in these protonemata (Fig. 3b), and found that V A and V B values were similar to each other, or V B values tended to be larger than V A values in some protonemata (10 out of 14 showed V A < V B ). Thus, effective Dendra2 movement in the apical direction seems to be eliminated in the non-growing protonemata.
Next, we examined the effect of a metabolic inhibitor, NaN3, on the movement in the growing protonemata under the red light. We determined V A and V B values of the protonemata that were treated with 30 μM NaN3 2 h before the start of time-lapse imaging (Fig. 3c). We found that the apical directional movement of Dendra2 was eliminated (6 out of 14 showed V A > V B ; 7 out of 14 showed V A < V B ; 1 out of 14 showed V A = V B ). The rate of the cell-to-cell movement of Dendra2 was not affected at 30 μM NaN3 (Fig. 3; V A [Red light] = 1.26 ± 0.63 min−1, V A [NaN3] = 1.15 ± 0.72 min−1; V B [Red light] = 0.87 ± 0.37 min−1, V B [NaN3] = 1.15 ± 0.71 min−1, mean ± SD). The growth of the protonemata was completely arrested within 2 h of the start of NaN3 treatment (Fig. S4). Higher concentrations of NaN3 (i.e., 1 and 10 mM) inhibited the cell-to-cell movement of Dendra2 (Fig. S5).
Long-term directional movement of Dendra2 along protonemata
We also observed the long-term Dendra2 movement along the protonemata. After time-lapse imaging was carried out (Fig. 3), we cultured the protonemata overnight in their respective conditions (red light, dark or NaN3). Then we observed the spread of Dendra2 fluorescence along the protonemal filament by fluorescence microscopy (Fig. 4). We measured the mean fluorescence intensities per pixel of Cell [+2], Cell [+1], Cell [0] (photoconverted cell), Cell [−1] and Cell [−2] (see Fig. 4). The average of the relative fluorescence intensity per pixel of each cell is shown in Fig. 4a (red light), b (dark) and c (NaN3). In the growing protonemata cultured under red light, we found that the relative fluorescence intensity of Cell [+1] was significantly higher than that of Cell [−1] (P < 0.01), and the relative fluorescence intensity of Cell [+2] was also significantly higher than that of Cell [−2] (P < 0.01) (Fig. 4a). In addition, the relative fluorescence intensity of Cell [+1] was almost the same as that of Cell [0]. These results suggest that Dendra2 preferentially moves in the apical direction. By contrast, we did not detect such preferential spread of Dendra2 along the protonemal filament in the dark or in the presence of NaN3 (Fig. 4b, c).

Differences in Dendra2 movement along the protonemal filament depending on light conditions and metabolic activity. Each protonema used for time-lapse imaging (Fig. 3) was subsequently cultured overnight in the respective treatment conditions (red light, dark or NaN3), and then Dendra2 movement along the protonemal filament was observed by fluorescence microscopy in the protonemata cultured under red light (a), in the dark (b) or in the presence of 30 μM NaN3 (c). A representative fluorescence image of a protonema is shown. Only red fluorescence is shown. Mean fluorescence intensity per pixel of a photoconverted cell (Cell [0]) and its surrounding cells (Cell [+2], [+1], [−1] and [−2]) were measured with ImageJ. For each cell, mean fluorescence intensity per pixel was normalized to that of Cell [0]. The average of the relative fluorescence intensity per pixel of each cell was graphically represented. Data represent the mean ± SD. n = 17 (red light), 12 (dark) and 12 (NaN3). Scale bars 100 μm. **P < 0.01
Discussion
In this study, we established a novel tool for analyzing cell-to-cell movement of macromolecule via PD at a single-cell level by using a simple filamentous tissue, moss protonemata, and a photoconvertible fluorescent protein, Dendra2. We then used this tool to find and measure acropetal transport between protonemal cells. Although such directional movement of molecules along the protonemal axes in mosses has been considered (Duckett et al. 1998), to the best of our knowledge, this is the first report to show that this transport occurs in moss protonemata.
Previously, PD transport was mostly studied in angiosperms, and in particular, for the analysis of non-targeted movement of molecules via PD at a single cell level, simple filamentous tissues such as multi-cellular trichomes (Christensen et al. 2009; Waigmann and Zambryski 2000) and stamen hairs (Radford and White 2011; Tucker 1982) have often been used. In stamen hairs, the regulation of PD transport has been quantitatively analyzed by calculating the rate of cell-to-cell movement of a fluorescent dye microinjected into a single cell (Tucker 1987, 1988, 1993; Tucker and Boss 1996; Tucker and Tucker 1993; Tucker et al. 1989). However, dye injection may affect PD transport by loss of cellular turgor pressure as a result of cellular wounding (Oparka and Prior 1992). In this study, Dendra2 was photoconverted non-invasively and can be used to mark any single cell at any time. Additionally, since fluorescent proteins including Dendra2 move between cells by diffusion as fluorescent dyes do (Crawford and Zambryski 2001), we can quantitatively and easily analyze non-targeted protein movement at the cellular level.
Thus, as shown in Fig. 2, we were able to measure the kinetics of Dendra2 movement between cells at intervals of at least every fifteen minutes. PD transport can be dynamically changed by biotic and abiotic stresses in angiosperms (Burch-Smith and Zambryski 2012); for instance, sucrose unloading in pea root tips is increased under osmotic stress, with the PD widening within a few hours (Schulz 1994, 1995). Our tool will enable us to quantitatively analyze such relatively rapid regulation of PD transport in response to environmental stresses.
Here we found the directional transport of a non-specific protein, Dendra2 via PD toward the apical direction along the protonemal filament (Figs. 2, 3). In targeted movement, in which some specific proteins actively increase the PD SEL to allow their passage into a neighboring cell, those proteins were directionally transported between cells toward their target cells (Wu and Gallagher 2011). By contrast, although non-targeted movement via PD represents diffusible movement of non-specific molecules, there have been only a few reports in which non-specific molecules such as fluorescent dyes and proteins can be directed. Christensen et al. (2009) reported the unidirectional transport of non-targeted molecules at the specific cell interface between epidermal and trichome cells in tobacco leaves. Although this interface has been implicated in the establishment of symplastic fields in trichomes (Waigmann and Zambryski 1995), it is unknown whether and how this directional transport affects development and function of the trichomes. The directional transport that we found in the moss protonemata was unlikely to result from a similar, strict regulation as found at the epidermal/trichome interface, because directional movement was detected in several different cells that we used between the 10th cell and more basal cells (Fig. 3a). In addition, we observed directional transport only in the growing protonemata, but not in non-growing protonemata incubated in the dark or in the presence of NaN3. It will be interesting to determine whether and how such directional transport via PD can affect protonemal growth and development. For instance, this directional transport via PD may promote the transport of nutrients and photosynthetic products toward the apical part of the protonemata to maintain their growth. Further experiments will be necessary to answer this question.
Our data showed that the directional transport along a protonemal filament was eliminated by incubation in the dark or treatment with NaN3, implying that directional transport requires the photosynthetic and metabolic activities of protonemal cells. In previous reports, sodium azide increased PD SEL while drastically reducing cellular ATP levels in a variety of tissues in angiosperms (Christensen et al. 2009; Cleland et al. 1994; Tucker 1993), suggesting that at least some aspects of PD function are under strict ATP-dependent control. Indeed, several proteins whose functions are ATP-dependent are localized to PD and considered to be regulators of PD transport (Ding et al. 1996; Fernandez-Calvino et al. 2011; Radford and White 2011). Although it is unknown whether such ATP-dependent regulation of PD functions in moss protonemata, it is possible that ATP-dependent, PD-localized proteins regulate not only PD SEL but also directional movement through PD. Another possibility is that mass flow may establish the directional movement in the protonemata, which is independent of PD regulation itself. In fungal hyphae, which form filamentous structures with a growing tip, pressure-driven mass flow transports solutes toward the apex across the septum (Lew 2005, 2011). It has been predicted that this pressure-driven mass flow is caused by osmotic imbalances along the hyphal filaments: imbalances that are established by the activity of H+-ATPase and various ion transporters (Lew 2011). These two pathways driving directional transport via PD may not be mutually exclusive in the protonemata.
We expect that studies of PD transport using moss protonemata may allow us to find novel aspects of PD regulation. For example, as shown in Fig. S5, higher concentrations of NaN3 inhibited the intercellular movement of Dendra2, whereas at the same concentrations, i.e. 1–10 mM NaN3, the treatment enhanced cell-to-cell diffusion in angiosperms (Christensen et al. 2009; Cleland et al. 1994; Tucker 1993), suggesting that depletion of ATP caused by azide treatment may lead to reduction of PD SEL or closure of PD in the moss but increase in angiosperms. This indicates differences in PD transport between moss and angiosperms. Furthermore, we found that intercellular movement of Dendra2 was very low in cells from apical to the 10th cell (materials and methods, data not shown), suggesting that PD SEL could be distinct depending on the cell position along the protonemata. Because protonemal cells can be differentiated from the apex to the base of protonemata (Pressel et al. 2008), the study of PD SEL along protonemal filaments may uncover when and how PD SEL could be regulated depending on the cellular status. Thus, the protonemata developed here will be a powerful tool for uncovering dynamic regulation of PD transport, which depends on environmental responses and differentiation states of cells, and also may lead to discovery of the unique mechanisms involved in regulation of PD transport, such as directional transport via PD, which we observed.
References
Abramoff MD, Magalhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42
Aoyama T, Hiwatashi Y, Shigyo M, Kofuji R, Kubo M, Ito M, Hasebe M (2012) AP2-type transcription factors determine stem cell identity in the moss Physcomitrella patens. Development 139:3120–3129
Ashton NW, Cove DJ (1977) Isolation and preliminary characterization of auxotrophic and analog resistant mutants of moss, Physcomitrella patens. Mol Gen Genet 154:87–95
Burch-Smith TM, Zambryski PC (2012) Plasmodesmata paradigm shift: regulation from without versus within. Annu Rev Plant Biol 63:239–260
Burch-Smith TM, Stonebloom S, Xu M, Zambryski PC (2011) Plasmodesmata during development: re-examination of the importance of primary, secondary, and branched plasmodesmata structure versus function. Protoplasma 248:61–74
Christensen NM, Faulkner C, Oparka K (2009) Evidence for unidirectional flow through plasmodesmata. Plant Physiol 150:96–104
Chudakov DM, Lukyanov S, Lukyanov KA (2007) Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc 2:2024–2032
Cleland RE, Fujiwara T, Lucas WJ (1994) Plasmodesmal-mediated cell-to-cell transport in wheat roots is modulated by anaerobic stress. Protoplasma 178:81–85
Cook ME, Graham LE, Botha CEJ, Lavin CA (1997) Comparative ultrastructure of plasmodesmata of Chara and selected bryophytes: toward an elucidation of the evolutionary origin of plant plasmodesmata. Am J Bot 84:1169–1178
Cove D, Bezanilla M, Harries P, Quatrano R (2006) Mosses as model systems for the study of metabolism and development. Annu Rev Plant Biol 57:497–520
Crawford KM, Zambryski PC (1999) Plasmodesmata signaling: many roles, sophisticated statutes. Curr Opin Plant Biol 2:382–387
Crawford KM, Zambryski PC (2001) Non-targeted and targeted protein movement through plasmodesmata in leaves in different developmental and physiological states. Plant Physiol 125:1802–1812
Dashevskaya S, Kopito RB, Friedman R, Elbaum M, Epel BL (2008) Diffusion of anionic and neutral GFP derivatives through plasmodesmata in epidermal cells of Nicotiana benthamiana. Protoplasma 234:13–23
Ding B, Kwon MO, Warnberg L (1996) Evidence that actin filaments are involved in controlling the permeability of plasmodesmata in tobacco mesophyll. Plant J 10:157–164
Duckett CM, Oparka KJ, Prior DAM, Dolan L, Roberts K (1994) Dye-coupling in the root epidermis of Arabidopsis is progressively reduced during development. Development 120:3247–3255
Duckett JG, Schmid AM, Ligrone R (1998) Protonemal morphogenesis. In: Bates JW, Ashton NW, Duckett JG (eds) Bryology for the twenty-first century. Maney, Leeds, pp 223–246
Ehlers K, van Bel AJ (2010) Dynamics of plasmodesmal connectivity in successive interfaces of the cambial zone. Planta 231:371–385
Epel BL (2009) Plant viruses spread by diffusion on ER-associated movement-protein-rafts through plasmodesmata gated by viral induced host β-1,3-glucanases. Semin Cell Dev Biol 20:1074–1081
Fernandez-Calvino L, Faulkner C, Walshaw J, Saalbach G, Bayer E, Benitez-Alfonso Y, Maule A (2011) Arabidopsis plasmodesmal proteome. PLoS ONE 6:13
Gisel A, Hempel FD, Barella S, Zambryski P (2002) Leaf-to-shoot apex movement of symplastic tracer is restricted coincident with flowering in Arabidopsis. Proc Natl Acad Sci USA 99:1713–1717
Imlau A, Truernit E, Sauer N (1999) Cell-to-cell and long-distance trafficking of the green fluorescent protein in the phloem and symplastic unloading of the protein into sink tissues. Plant Cell 11:309–322
Kim I, Zambryski PC (2005) Cell-to-cell communication via plasmodesmata during Arabidopsis embryogenesis. Curr Opin Plant Biol 8:593–599
Kim I, Kobayashi K, Cho E, Zambryski PC (2005) Subdomains for transport via plasmodesmata corresponding to the apical-basal axis are established during Arabidopsis embryogenesis. Proc Natl Acad Sci USA 102:11945–11950
Kobayashi K, Kim I, Cho E, Zambryski P (2005) Plasmodesmata and plant morphogenesis. In: Oparka KJ (ed) Plasmodesmata. Annual plant reviews, vol. 18. Blackwell, Oxford, pp 90–112
Laemmli UK (1970) Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature 227:680–685
Lew RR (2005) Mass flow and pressure-driven hyphal extension in Neurospora crassa. Microbiology-(UK) 151:2685–2692
Lew RR (2011) How does a hypha grow? The biophysics of pressurized growth in fungi. Nat Rev Microbiol 9:509–518
Liarzi O, Epel BL (2005) Development of a quantitative tool for measuring changes in the coefficient of conductivity of plasmodesmata induced by developmental, biotic, and abiotic signals. Protoplasma 225:67–76
Liesche J, Schulz A (2012a) In vivo quantification of cell coupling in plants with different phloem-loading strategies. Plant Physiol 159:355–365
Liesche J, Schulz A (2012b) Quantification of plant cell coupling with three-dimensional photoactivation microscopy. J Microsc 247:2–9
Martens HJ, Hansen M, Schulz A (2004) Caged probes: a novel tool in studying symplasmic transport in plant tissues. Protoplasma 223:63–66
Maule AJ (2008) Plasmodesmata: structure, function and biogenesis. Curr Opin Plant Biol 11:680–686
Nishiyama T, Hiwatashi Y, Sakakibara K, Kato M, Hasebe M (2000) Tagged mutagenesis and gene-trap in the moss, Physcomitrella patens by shuttle mutagenesis. DNA Res 7:9–17
Oparka K, Boevink P (2005) Techniques for imaging intercellular transport. In: Oparka KJ (ed) Plasmodesmata annual plant reviews, vol 18. Blackwell, Oxford, pp 241–262
Oparka KJ, Prior DAM (1992) Direct evidence for pressure-generated closure of plasmodesmata. Plant J 2:741–750
Oparka KJ, Roberts AG, Boevink P, Santa Cruz S, Roberts L, Pradel KS, Imlau A, Kotlizky G, Sauer N, Epel B (1999) Simple, but not branched, plasmodesmata allow the nonspecific trafficking of proteins in developing tobacco leaves. Cell 97:743–754
Palevitz BA, Hepler PK (1985) Changes in dye coupling of stomatal cells of allium and commelina demonstrated by microinjection of Lucifer yellow. Planta 164:473–479
Pressel S, Ligrone R, Duckett JG (2008) Cellular differentiation in moss protonemata: a morphological and experimental study. Ann Bot 102:227–245
Radford JE, White RG (2011) Inhibitors of myosin, but not actin, alter transport through Tradescantia plasmodesmata. Protoplasma 248:205–216
Roberts AG, Oparka KJ (2003) Plasmodesmata and the control of symplastic transport. Plant Cell Environ 26:103–124
Ruan YL, Llewellyn DJ, Furbank RT (2001) The control of single-celled cotton fiber elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansion. Plant Cell 13:47–60
Rydin H, Clymo RS (1989) Transport of carbon and phosphorus-compounds about Sphagnum. Proc R Soc B-Biol Sci 237:63–84
Schnepf E, Reinhard C (1997) Brachycytes in Funaria protonemate: induction by abscisic acid and fine structure. J Plant Physiol 151:166–175
Schnepf E, Sawidis T (1991) Filament disruption in Funaria protonemata: occlusion of plasmodesmata. Bot Acta 104:98–102
Schulz A (1994) Phloem transport and differential unloading in pea seedlings after source and sink manipulations. Planta 192:239–248
Schulz A (1995) Plasmodesmal widening accompanies the short-term increase in symplastic phloem unloading in pea root-tips under osmotic stress. Protoplasma 188:22–37
Tucker EB (1982) Translocation in the staminal hairs of Setcreasea purpurea. I. A study of cell ultrastructure and cell-to-cell passage of molecular probes. Protoplasma 113:193–201
Tucker EB (1987) Cytoplasmic streaming does not drive intercellular passage in staminal hairs of Setcreasea purpurea. Protoplasma 137:140–144
Tucker EB (1988) Inositol bisphosphate and inositol trisphosphate inhibit cell-to-cell passage of carboxyfluorescein in staminal hairs of Setcreasea purpurea. Planta 174:358–363
Tucker EB (1993) Azide treatment enhances cell-to-cell diffusion in staminal hairs of Setcreasea purpurea. Protoplasma 174:45–49
Tucker EB, Boss WF (1996) Mastoparan-induced intracellular Ca2+ fluxes may regulate cell-to-cell communication in plants. Plant Physiol 111:459–467
Tucker EB, Tucker JE (1993) Cell-to-cell diffusion selectivity in staminal hairs of Setcreasea purpurea. Protoplasma 174:36–44
Tucker JE, Mauzerall D, Tucker EB (1989) Symplastic transport of carboxyfluorescein in staminal hairs of Setcreasea purpurea is diffusive and includes loss to the vacuole. Plant Physiol 90:1143–1147
Waigmann E, Zambryski P (1995) Tobacco mosaic virus movement protein-mediated protein transport between trichome cells. Plant Cell 7:2069–2079
Waigmann E, Zambryski P (2000) Trichome plasmodesmata: a model system for cell-to-cell movement. Adv Bot Res 31:261–283
Wu S, Gallagher KL (2011) Mobile protein signals in plant development. Curr Opin Plant Biol 14:563–570
Wu XL, Dinneny JR, Crawford KM, Rhee Y, Citovsky V, Zambryski PC, Weigel D (2003) Modes of intercellular transcription factor movement in the Arabidopsis apex. Development 130:3735–3745
Zambryski P, Crawford K (2000) Plasmodesmata: gatekeepers for cell-to-cell transport of developmental signals in plants. Annu Rev Cell Dev Biol 16:393–421
Acknowledgments
We thank Dr. Konstantin A. Lukyanov for providing the vector pDendra2-C. We thank Drs. Minoru Kubo and Mitsuyasu Hasebe for providing the vector pT1OG. We thank Mr. Hiroya Funakawa for his support in making transgenic plants. We thank Dr. Natsumi Noda for her support in statistical analysis. We also thank Drs. Kotaro Yamamoto, Masaaki Watahiki and Mr. Liang Bao for valuable discussions. This work was supported, in part, by grants from the Ministry of Education, Science, Culture, and Sports of Japan and the Japan Society for the Promotion of Science to T.F. M.K. was the recipient of a Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science, and a Grants-in-Aid for Scientific Research for Plant Graduate Students from Nara Institute of Science and Technology, the Ministry of Education, Culture, Sports, Science, and Technology. The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kitagawa, M., Fujita, T. Quantitative imaging of directional transport through plasmodesmata in moss protonemata via single-cell photoconversion of Dendra2. J Plant Res 126, 577–585 (2013). https://doi.org/10.1007/s10265-013-0547-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-013-0547-5