
Abstract
The sensory hair cells of the cochlea and vestibular organs are essential for normal hearing and balance function. The mammalian ear possesses a very limited ability to regenerate hair cells and their loss can lead to permanent sensory impairment. In contrast, hair cells in the avian ear are quickly regenerated after acoustic trauma or ototoxic injury. The very different regenerative abilities of the avian vs. mammalian ear can be attributed to differences in injury-evoked expression of genes that either promote or inhibit the production of new hair cells. Gene expression is regulated both by the binding of cis-regulatory molecules to promoter regions as well as through structural modifications of chromatin (e.g., methylation and acetylation). This study examined effects of histone deacetylases (HDACs), whose main function is to modify histone acetylation, on the regulation of regenerative proliferation in the chick utricle. Cultures of regenerating utricles and dissociated cells from the utricular sensory epithelia were treated with the HDAC inhibitors valproic acid, trichostatin A, sodium butyrate, and MS-275. All of these molecules prevent the enzymatic removal of acetyl groups from histones, thus maintaining nuclear chromatin in a “relaxed” (open) configuration. Treatment with all inhibitors resulted in comparable decreases in supporting cell proliferation. We also observed that treatment with the HDAC1-, 2-, and 3-specific inhibitor MS-275 was sufficient to reduce proliferation and that two class I HDACs—HDAC1 and HDAC2—were expressed in the sensory epithelium of the utricle. These results suggest that inhibition of specific type I HDACs is sufficient to prevent cell cycle entry in supporting cells. Notably, treatment with HDAC inhibitors did not affect the differentiation of replacement hair cells. We conclude that histone deacetylation is a positive regulator of regenerative proliferation but is not critical for avian hair cell differentiation.
Similar content being viewed by others

Introduction
Sensory hair cells are mechanoreceptors that are responsible for the detection of sound vibrations (in the cochlea) and head movements (in the vestibular organs). Hair cells are produced during embryonic development and can be lost later in life as a consequence of treatment with ototoxic drugs, noise exposure, or as part of the aging process. Because the mammalian inner ear has a very limited ability to repair or regenerate hair cells, the loss of these cells can lead to permanent hearing loss and/or balance disorders. In contrast, the ears of nonmammalian vertebrates can quickly regenerate hair cells after noise trauma or ototoxic injury (Corwin and Oberholtzer 1997). Microarray-based studies have identified numerous changes in gene expression that occur during sensory regeneration in the avian ear (Hawkins et al. 2003, 2007). Similar changes in transcriptional activity have been demonstrated in the developing mammalian inner ear, suggesting certain commonalities between development and regeneration (Sajan et al. 2007). A more complete understanding of changes in gene expression during regeneration and the regulatory mechanisms that dictate these changes will be valuable in the development of methods for inducing sensory regeneration in the human inner ear.
Gene expression is regulated both by the presence and activity of transcription factors and by epigenetic mechanisms such as the posttranslational modification of histones (Jenuwein and Allis 2001; Li et al. 2007). Acetylation of histones causes a relaxation of DNA coiling from histone cores, allowing for transcriptional complexes and other factors to interact with regulatory regions (Kouzarides 2007). Early studies have demonstrated that treatment with sodium butyrate can cause hyperacetylation of histones, by inhibiting enzymes known as histone deacetylases (HDACs). Treatment with HDAC inhibitors can lead to a variety of effects on cultured cells, including decreased proliferation and prodifferentiation (Kruh 1982). Although the principal role of HDACs is to reduce the acetylation of histones, it has also been shown that HDACs can affect gene transcription through deacetylation of transcription factors (Bolden et al. 2006). Numerous other compounds have subsequently been discovered that inhibit HDACs. These include the antiseizure drug valproic acid (VPA), trichostatin A (TSA—a natural product from Streptomyces), and the synthetic molecule MS-275 (Phiel et al. 2001; Saito et al. 1999; Yoshida et al. 1990). HDACs are categorized into four classes, with MS-275 acting on class I HDACs (with the exception of HDAC8) and VPA and sodium butyrate (NaB) inhibiting classes I and IIa. In contrast, TSA has a broad inhibition profile and acts on classes I, IIa, IIb, and IV HDACs (Bolden et al. 2006; Gottlicher et al. 2001; Khan et al. 2008). HDAC inhibitors have been intensively studied in tumor biology and lead to cycle arrest and increased differentiation in a wide variety of malignant cell lines (Carey and La Thangue 2006; Vigushin and Coombes 2002). Inhibition of HDACs can also influence cells in the developing nervous system, resulting in decreased proliferation, decreased differentiation of oligodendrocytes, and increased neuronal differentiation of both adult and embryonic neural stem cells (Balasubramaniyan et al. 2006; Hsieh and Gage 2004; Hsieh et al. 2004; Kondo 2006). Recently, it has been shown that supporting cell proliferation in the early-postnatal mouse utricle is decreased in response to treatment with NaB (Lu and Corwin 2008). Furthermore, HDACs have been described in the chicken, with TSA treatment shown to cause decreased HDAC1 and HDAC2 activity (Takami et al. 1999). In a separate study, it has been shown that angiogenesis is decreased in both mouse and chicken models following VPA treatment (Michaelis et al. 2004).
In the present study, we examine the regulatory effects of histone deacetylases on cell proliferation and differentiation in regenerating sensory epithelia from the avian ear, using four biochemically distinct inhibitors. We show that HDAC inhibition causes decreased proliferation of supporting cells in both dissociated epithelial cultures and organotypic cultures of the chick utricle. We also demonstrate that treatment with high concentrations of these inhibitors leads to apoptosis of supporting cells. Finally, we show that HDAC inhibition during vestibular regeneration does not lead to changes in cell fate.
Methods
Animals
White Leghorn chickens were hatched from fertile eggs (Charles River, Franklin, CT, USA) and housed in heated brooders within the animal care facility of Washington University. All animal-related protocols were approved by Washington University Institutional Animal Research Committee.
Culture techniques
Isolated sensory epithelia
Cultures of utricular sensory epithelia were prepared as previously described (Warchol 1995, 1999, 2002). Briefly, 7–14-day-old chicks were euthanized via CO2 asphyxiation. Following decapitation, the skin and mandible were removed and heads were placed in 70% EtOH. Utricles were quickly explanted from the temporal bones and placed in sterile Medium 199 with Hank’s salts and HEPES buffer. Following removal of the otoconia, utricles were incubated in thermolysin (500 μg/ml in Medium-199) for 1 h at 37°C and then returned to chilled medium for further dissection. A 30-gauge needle was used to remove sensory epithelium from the underlying basement membrane and associated connective tissue. The peripheral transitional epithelium was removed from the sensory epithelium with scissors and discarded. Isolated sheets of sensory epithelia were then cut into small fragments, incubated for 15 min in 0.05% trypsin at 37°C, and then triturated ten times in Medium 199 with 10% fetal bovine serum (FBS). Dissociated cells, consisting mostly of supporting cells, were plated onto laminin-coated culture wells (MatTek, Ashland, MA, USA). Each well contained cells from approximately one sensory epithelium, in a final volume of 50 μl. The medium for epithelial cultures consisted of Medium 199 with Earle’s salts 2,200 mg/l sodium bicarbonate, 0.69 mM l-glutamine, 25 mM HEPES, supplemented with 10% FBS. Cultures were first incubated overnight in 50 μl of medium, in order to allow cells to adhere to the laminin substrate. The next day, an additional 50 μl of medium was added to the cultures, for a final volume of 100 μl. Culture medium was replaced with fresh medium at 2-day intervals.
Preparation of organ cultures
Organ cultures of the chick utricle were prepared following previously described methods (Matsui et al. 2002). Briefly, utricles were dissected from chicks and placed in sterile Medium 199 with Hank’s salts and HEPES buffer. The otoconia were removed with fine forceps and the utricles were placed in MatTek wells with 100 μl of Medium 199 with Earle’s salts 2,200 mg/l sodium bicarbonate, 0.69 mM l-glutamine, 25 mM HEPES, supplemented with 1.0–2.5% FBS.
Pharmacological treatment of epithelial cultures
Epithelial cells were initially allowed to proliferate in vitro for 7 days at 37°C. Cultures were then incubated for either 24 or 48 h with either: NaB (1 mM), VPA (1 mM), MS-275 (5 μM), or TSA (100 nM; all obtained from Sigma, St. Louis, MO, USA). Doses were based on previously reported data from studies of cultured cells (Hsieh et al. 2004; Nishioka et al. 2008). Dose–response data were obtained by treating cultures for 24 h with either VPA (0.25, 0.5, and 1 mM), TSA (25, 50, and 100 nM), and MS-275 (1, 5, and 10 μM). In all experiments, control cultures were maintained in parallel but without HDAC inhibitor treatment. Control specimens for TSA and MS-275 contained 0.1% dimethyl sulfoxide (vehicle).
Pharmacological treatment of cultured utricles
Cultured utricles were incubated for 24 h in streptomycin sulfate (1 mM), in order to kill hair cells. Concentrations of streptomycin in this range have been shown to cause a near-complete loss of hair cells in both the striolar and extrastriolar regions of the utricle (Matsui et al. 2000). The cultures were then rinsed three times with fresh media and allowed to incubate for another 24 h in a streptomycin-free environment. At this point, cultures were treated with HDAC inhibitors and incubated for an additional 24 h. Control cultures (i.e., without HDAC inhibitors) were maintained concurrently. Proliferating cells in these cultures were labeled adding bromodeoxyuridine (BrdU; 10 μg/ml) to the medium for the final 4 h in vitro. Other utricles were treated with VPA (1 mM) for 8 days following streptomycin treatment, in order to determine the effects of ongoing HDAC inhibition on hair cell recovery.
Additional experiments examined the effects of HDAC inhibition specifically on hair cell differentiation during regeneration. In those studies, utricles were treated with 1 mM streptomycin for 24 h, rinsed thoroughly, and then maintained in drug-free media for 4 days. At this point, utricles were cultured with VPA (1 mM) for an additional 4 days. An equal number of control cultures were maintained in parallel but did not receive HDAC inhibitors. Finally, in order to assess for potential otoprotection by HDAC inhibitors, some utricles were given VPA (1 mM) simultaneously with streptomycin (1 mM) for 24 h, rinsed, and maintained in 1 mM VPA for an additional 24 h. Control utricles in those studies were treated with streptomycin alone.
Calretinin and HCS-1 immunohistochemistry
Cultures were fixed for 20 min in 4% paraformaldehyde (in 0.1-M phosphate buffer, pH 7.4). Subsequent histological processing was carried out at room temperature, unless otherwise noted. Specimens were first incubated for 2 h in blocking solution (phosphate-buffered saline (PBS), with 5% normal horse serum (NHS) and 0.2% Triton X-100), followed by overnight incubation in primary antibodies (at 4°C). The primary antibodies used in this study were: (1) anticalretinin (1:1,000, rabbit polyclonal, Calbiochem, La Jolla, CA, USA) and (2) HCS-1, a mouse monoclonal antibody that recognizes otoferlin in nonmammalian hair cells (Cyr et al. 2006; a gift from Dr. Jeffrey T. Corwin, University of Virginia). Secondary labeling for HCS-1 was carried out with a donkey antimouse antibody conjugated with Alexa Fluor 488 (Invitrogen, 1:500), while secondary labeling for calretinin was performed with a goat antirabbit antibody conjugated with Cy3 (GE Healthcare, Piscataway, NJ, USA; 1:500).
Bromodeoxyuridine immunohistochemistry
Proliferating supporting cells were labeled by adding BrdU (Sigma, 10 μg/ml) to cultures for the final 4 h in vitro. After fixation, DNA was denatured by treatment with HCl (epithelial cultures: 1 N for 15 min; whole utricles: 2 N HCl for 30 min). Specimens were then rinsed with PBS and incubated in blocking solution (as above), followed by overnight incubation in mouse monoclonal anti-BrdU (Becton Dickinson, San Jose, CA, USA, 1:50, in PBS with 2% NHS and 0.1% Triton X-100) at 4°C, and then 2-h incubation in donkey antimouse antibody, conjugated to Alexa Fluor 488 (1:500; Invitrogen).
Fluorescent staining of nucleic acid
All specimens were stained with 4′,6-diamidino-2-phenyindole (DAPI; Sigma-Aldrich, 2.67 μM) for 2 h, in order to label cell nuclei. Utricles were then mounted on glass slides in 90% glycerol/10% PBS, while epithelial cultures were coverslipped directly in MatTek dishes, using the same mounting medium.
RT-PCR detection of HDAC1 and HDAC2
Dissociated epithelial cultures were triturated in 100 μl per well of TRIzol Reagent (Invitrogen) and precipitated with isopropanol. Total RNA was used to create complementary DNA with RETROscript kit (Ambion, Austin, TX, USA), following manufacturer’s instructions. Polymerase chain reaction (PCR) was performed using Illustra Ready-To-Go reverse transcription (RT)-PCR Beads (GE Healthcare). HDAC1 forward and reverse primer sequences were 5’ cggatgacccacaacctact 3’ and 5’ tccagcagagagctgacaga 3’. HDAC2 forward and reverse primers were 5’ taccgaccccacaaagctac 3’ and 5’ cagccatgtctgtctgctgt 3’. Expected amplification sizes were 240 and 238 bp for HDAC1 and HDAC2, respectively.
Data analysis
Quantification of DAPI- and BrdU-labeled nuclei from epithelial cultures.
Labeled epithelial cultures were visualized on an inverted microscope (Nikon Eclipse 2000) with epifluorescent illumination. Images were captured using a cooled charge-coupled device camera (Q Imaging) and stored as .tiff files. Each specimen was viewed with a ×20 objective and images of both BrdU-labeled and DAPI-labeled nuclei were obtained from five randomly selected 138,195-μm2 regions (with a density of approximately 200 nuclei per field). The numbers of DAPI- and BrdU-labeled cells were quantified using Image-Pro software (Media Cybernetics; Bethesda, MD, USA) and counts were normalized to 10,000 μm2. These data were then used to calculate proliferation indices, defined as the number of BrdU-labeled cells/DAPI-labeled cells per image field. Similarly, the apoptosis index (number of pyknotic nuclei/total number of DAPI-stained nuclei) was calculated for randomly selected regions. Three sets of cultures were replicated for all experiments, except for dose–response experiments, which consisted of two replications each.
Quantification of BrdU-, HCS-1-, or calretinin-labeled cells from cultured utricles.
Cell proliferation in intact utricles was quantified by imaging BrdU-labeled cells in four randomly selected extrastriolar regions per utricle, using a ×40 objective (size of sampled field = 37,714 μm2). Since the surface area of the chick utricle is about 1.52 mm2 (Warchol 2001), our samples represented about 10% of the entire sensory epithelium. Similarly, HCS-1- or calretinin-labeled cells were imaged in four to five extrastriolar fields per utricle (using a ×60 objective), which represented approximately 5% of the specimen’s surface area. The numbers of BrdU-, HCS-1-, or calretinin-labeled cells per field were counted from stored images and normalized to per 10,000 μm2. For each experiment, data were obtained from three individual sets of cultures.
Statistics.
Data were analyzed using either an unpaired two-sample t test with unequal variances (Microsoft Excel, Microsoft, Redmond, WA, USA) or analysis of variance (ANOVA) with multiple comparisons (Tukey test) with SigmaStat (Systat, San Jose, CA, USA). Nonparametric data were analyzed using the Kruskal–Wallis ANOVA (SigmaStat), with multiple comparisons. All data were expressed as mean ± standard deviation.
Results
Effects of HDAC inhibitors on chick inner ear cultures
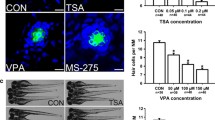
Supporting cells from the avian utricle continue to proliferate at high levels when maintained in primary culture (Warchol 1995, 2002). In order to determine the effect of histone acetylation on supporting cell division, cultures of dissociated utricular supporting cells were treated with HDAC inhibitors and proliferating cells were identified by the incorporation of BrdU (Fig. 1). Treatment for 24 h with the HDAC inhibitors VPA (1 mM), TSA (100 nM), or NaB (1 mM) resulted in an 85–95% reduction in supporting cell proliferation (Fig. 2A). Comparable results were observed following 48 h treatments with these compounds (data not shown). Treatment with VPA was also found to reduce supporting cell proliferation in a dose-dependent fashion (Fig. 2B).

Decreased proliferation in epithelial cultures after treatment with VPA. Cultures of dissociated utricular supporting cells were treated for 24 h with 1 mM VPA and proliferating cells were labeled by the addition of BrdU for the final 4 h in vitro. Cultures were stained with DAPI (blue, left) and immunolabeled for BrdU (green, left and right). Shown are representative images of A VPA-treated and B control cultures. Scale bar 100 μm.

Quantification of proliferation in epithelial cultures following treatment with HDAC inhibitors. Cultures were treated for 24 h with VPA (1 mM), TSA (100 nM), or NaB (1 mM). Following fixation and histological processing, proliferation indices (BrdU-labeled cells/total cells per 10,000 μm2) were quantified for randomly selected image fields. A Cultures treated for 24 h with all HDAC inhibitors displayed >80% reduction in proliferation. B Dose–response relationship for treatment with VPA. Dissociated cultures were treated at varying doses (0, 0.25, 0.5, and 1 mM) for 24 h, and BrdU was added for the final 4 h in vitro. Increasing concentrations of VPA caused dose-dependent decreases in the proliferation index. Error bars: standard deviation, *p < 0.001.
Given that treatment with HDAC inhibitors reduced the proliferation of dissociated supporting cells, we next examined the effects of HDAC inhibitors on regeneration in the intact chick utricle. Utricles were placed in organotypic culture and hair cells were lesioned by adding streptomycin (1 mM) to the medium for the first 24 h in vitro. Initial experiments quantified the resulting hair cell lesion, both with and without VPA treatment. Utricles (n = 16) were cultured in 1 mM streptomycin; half of these cultures also received 1 mM VPA. After 24 h, the specimens were rinsed. Specimens that received VPA were maintained for another 24 h with VPA, while control utricles were maintained in a drug-free medium. Following fixation, surviving hair cells were immunolabeled with the HCS-1 antibody and quantified in the extrastriolar regions of each specimen. Utricles that received streptomycin contained 3.1 ± 4.0 HCs per 10,000 μm2, while utricles that received streptomycin + VPA contained 3.0 ± 3.3 HCs per 10,000 μm2. In contrast, untreated utricles (n = 4) had 103.4 ± 22.2 HCs per 10,000 μm2. These data demonstrate that streptomycin treatment resulted in a significant loss of hair cells and that ototoxic effects of streptomycin were not blocked by cotreatment with VPA (Fig. 3).

Treatment with VPA does not protect hair cells from streptomycin ototoxicity. Organotypic cultures of the chick utricle were used to determine whether VPA treatment reduced the ototoxic effects of streptomycin. Utricles were cultured for 48 h in A control medium, B 24 h in 1 mM streptomycin followed by 24 h in drug-free control medium, or C 24 h in 1 mM streptomycin and 1 mM VPA followed by 24 h with 1 mM VPA alone. Utricles were then fixed and surviving hair cells were labeled with the HCS-1 antibody. Co-treatment with VPA did not appear to prevent hair cell death following streptomycin exposure. Scale bar 25 μm.
We then examined the effect of HDAC inhibitors on regenerative proliferation in the intact utricle. Prior data indicate that treatment in 1 mM streptomycin results in a small reduction in supporting cell proliferation at 24-h recovery, followed by a highly elevated level of proliferation at 48-h recovery (Matsui et al. 2000). For this reason, specimens were treated with HDAC inhibitors for 24 h, beginning at 24 h after streptomycin treatment (e.g., during hours 24–48 after streptomycin, the period during which cells enter S phase) and proliferating cells were labeled by addition of BrdU to the culture medium for the final 4 h in vitro. We found that treatment for 24 h with 1 mM VPA resulted in a 71% reduction in BrdU-labeled cells (control 21.4 ± 1.2 BrdU+ cells per 10,000 μm2, n = 8 vs. VPA-treated 6.2 ± 0.47 BrdU+ cells per 10,000 μm2, n = 9; p < 0.001; Fig. 4). Similarly, treatment with 1 mM NaB caused a 74% reduction in BrdU-labeled cells (control 16.2 ± 0.9 BrdU+ cells per 10,000 μm2 vs. NaB 4.2 ± 0.4 BrdU+ cells per 10,000 μm2, n = 8/8; p < 0.001). Thus, HDAC inhibition results in decreased supporting cell proliferation in both dissociated and organotypic cultures.

Treatment with VPA leads to decreased regenerative proliferation in cultured utricles. Chick utricles were placed in organotypic culture and treated for 24 h with 1 mM streptomycin. Specimens were then rinsed, maintained for 24 h in drug-free medium, followed by another 24 h in 1 mM VPA. Control cultures were treated identically but did not receive VPA. Proliferating cells were labeled by the addition of BrdU for the final 4 h in vitro. Control utricle A contains numerous BrdU-labeled cells (green), while the VPA-treated utricle B contains many fewer proliferating cells. C Quantification of BrdU-labeled cells per 10,000 μm2 shows >70% reduction in proliferation following VPA treatment. Error bars standard deviation, scale bar 100 μm, *p < 0.001.
Finally, we examined the influence of long-term HDAC inhibition on the recovery of hair cells in this culture system. Utricles were treated for 24 h with 1 mM streptomycin and were then allowed to recover in culture for 8 days. One group of specimens (n = 10) was treated with 1 mM VPA for the entire recovery period, while an equal number of specimens were maintained in normal medium. Regenerated hair cells were identified by immunoreactivity for HCS-1 and labeled cells in the extrastriolar regions were quantified. Utricles that were treated with VPA contained 23.88 ± 1.06 HCs per 10,000 μm2, while control utricles contained 53.40 ± 1.43 HCs per 10,000 μm2 (p < 0.0001; Fig. 5). This result indicated that HDAC inhibition for the entire recovery period interferes with hair cell regeneration.

Treatment with VPA for the entire recovery period causes decreased numbers of regenerated hair cells. Chick utricles (n = 20) were placed in culture and treated for 24 h with 1 mM streptomycin. They were then rinsed and maintained in vitro for an additional 8 days. Some cultures (n = 10) also received 1 mM VPA for the entire recovery period. Following fixation, hair cells were labeled with the HCS-1 antibody and quantified from randomly selected 10,000-μm2 portions of the extrastriolar regions. Normal hair cell recovery was observed in untreated utricles (A). Treatment with VPA for the entire regenerative period resulted in a 55% reduction in HCS-1-labeled cells (B, C). Error bars standard deviation, scale bar 25 μm, *p < 0.001.
Expression of HDAC1 and 2 in cultured sensory epithelia
Class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are constitutively expressed in most tissues and are inhibited by VPA, NaB, and TSA (Bolden et al. 2006). We verified the transcription of HDAC1 and HDAC2 in cultures of dissociated sensory epithelia using RT-PCR. Primers were designed from known mRNA sequences for both transcripts with expected product sizes of 240 bp for HDAC1 and 238 bp for HDAC2. Amplified products of the correct sizes were observed following RT-PCR (Fig. 6), indicating that HDAC1 and HDAC2 are present in the sensory epithelium of the chick utricle.

Expression of HDAC1 and HDAC2 in the sensory epithelium of the chick utricle. Cultures of dissociated supporting cells were prepared and mRNA extracted with TRIzol. Reverse-transcription polymerase chain reaction was performed, using primers for HDAC1 (lane 2) and HDAC2 (lane 4). The expected fragment size was amplified for both targets. Lanes 1 and 6 contain 1-kb ladder, lanes 3 and 5 are HDAC1 and HDAC2 negative controls, respectively.
Specific HDAC inhibition
As noted above, structural and pharmacological studies of HDAC inhibitors have identified several distinct chemical families that have the ability to inhibit different HDACs (Bolden et al. 2006). To investigate the possible involvement of specific HDAC classes on supporting cell proliferation, we next examined the effects of a more specific HDAC inhibitor on dissociated utricular cells. Previous studies have shown that the compound MS-275 selectively inhibits most class I HDACs. Specifically, treatment with MS-275 has a strong inhibitory effect on HDAC1 and HDAC2, a moderate inhibitory effect on HDAC3, and little or no activity against other HDAC classes (Hu et al. 2003). Treatment with MS-275 at three doses (1, 5, and 10 μM) reduced supporting cell proliferation by 58–80% (Fig. 7A). We also treated cultures with MS-275 in combination with TSA (a pan-class HDAC inhibitor). The reduction in proliferation caused by application of both inhibitors was similar to that caused by either inhibitor applied alone (Fig. 7B), suggesting that inhibition of HDAC1, 2, and 3 are sufficient for the observed antiproliferative effects.

HDAC1, 2, and 3 inhibition causes decreased proliferation. A Dissociated supporting cell cultures were treated for 24 h with various doses of MS-275, an HDAC 1, 2, and 3 specific inhibitor. We observed that MS-275 treatment caused a dose-dependent decrease in proliferation. B Dissociated cultures treated with increasing doses of TSA. A comparable decrease of proliferation occurred with both 50- and 100-nM doses. C Dissociated cultures were treated with either TSA (100 nM), MS-275 (5 μm), or both drugs, and proliferation indices were calculated. No change in proliferation occurred between treated groups. Error bars: standard deviation, *p < 0.05.
High concentrations of HDAC inhibitors induce apoptosis
Activation of HDACs has been shown to play a role in both cell proliferation and in programmed cell death (Carey and La Thangue 2006). In the previous experiments, we observed numerous pyknotic nuclei in cultures that had been treated with both TSA and MS-275 (e.g., Fig. 8A). Pyknosis is a hallmark of apoptosis (Kerr et al. 1972). In order to assess the levels of cell death induced by HDAC inhibition, we quantified the relative percentage of pyknotic nuclei vs. total DAPI-stained nuclei in randomly selected regions of epithelial cultures. Cultures treated with TSA/MS-275 showed a >10-fold increase in apoptosis, compared to control cultures (p < 0.05; Fig. 8B). Cultures treated with either MS-275 or TSA alone also contained more apoptotic nuclei than did controls but significantly less than combined TSA/MS-275 treatment (p < 0.05; Fig. 8B). We also observed increased cleaved caspase 3 immunolabeling following TSA (100 nM) treatment but not after VPA (1 mM) treatment (Fig. 8C–E). In order to quantify the relative toxicity of different HDAC inhibitors, dissociated epithelial cultures were treated for 24 h with either: VPA (1 mM), NaB (1 mM), or TSA (100 nM). Treatment with TSA resulted in the highest levels of apoptosis, while NaB caused a more modest increase in apoptosis (Fig. 9A). Notably, treatment with 1 mM VPA did not cause a statistically significant increase in apoptosis over control levels. Dose–response relationships for MS-275 and TSA revealed that the proapoptotic effects occurred at and beyond the concentrations that yielded maximal antiproliferative effects (Fig. 9B, C).

Treatment with both TSA and MS-275 causes increased apoptosis. A Dissociated supporting cells treated with both TSA (100 nM) and MS-275 (5 μM) contained increased numbers of pyknotic nuclei (arrow), scale bar 25 μm. B Dissociated cultures were treated for 24 h with either MS-275 (5 μM), TSA (100 μM), or both, and the resulting apoptosis indices (# pyknotic nuclei/# DAPI-stained nuclei per 10,000 μm2) were calculated. Simultaneous treatment with both inhibitors caused a large increase in apoptosis. Immunoreactivity for cleaved caspase 3 (red) was examined in untreated dissociated supporting cells (C), as well as cells treated with either 1 mM VPA for 24 h (D) or 100 nM TSA for 24 h (E). A dramatic increase in cleaved caspase 3 was noted following TSA treatment but not after VPA treatment. Nuclei stained with DAPI (blue). Scale bar 100 μm, error bars: standard deviation, *p < 0.05.

Quantification of apoptosis induced by high concentrations of HDAC inhibitors. A Dissociated supporting cells were treated for 24 h with 1 mM NaB, 1 mM VPA, 5 μM MS-275, or 100 nM TSA. Following fixation, cell nuclei were stained with DAPI. Apoptosis indices (number of pyknotic nuclei/total DAPI-stained nuclei per 10,000 μm2) were calculated for all three HDAC inhibitors. Dose-dependant increases in apoptosis were observed after treatment with MS-275 (B), and TSA (C). Error bars: standard deviation, *p < 0.05.
HDAC inhibitor effects on hair cell differentiation
Prior studies have shown that treatment with HDAC inhibitors causes neural stem cells to differentiate as neurons, astrocytes, and oligodendrocytes (Hsieh and Gage 2004; Kondo 2006). In order to determine whether a similar effect might occur with inner ear supporting cells, we examined the effects of HDAC inhibition on the differentiation of hair cells during sensory regeneration. Utricles were placed in culture and treated for 24 h with 1 mM streptomycin. They were then rinsed and maintained in drug-free media for 4 days, in order to allow time for regenerative proliferation (Matsui et al. 2000). At this point, cultures (n = 8) received 1 mM VPA for 4 days and were then fixed and processed for immunocytochemical labeling of regenerated hair cells. An equal number of utricles were cultured in parallel but did not receive VPA; these specimens served as controls. We used two markers of hair cell phenotype—the HCS-1 antibody (Cyr et al. 2006) and an antibody against the calcium binding protein calretinin (Rogers 1989). Quantification of both HCS-1- and calretinin-labeled cells revealed no differences in the numbers of regenerated hair cells in VPA-treated utricles, when compared with control specimens (Fig. 10). We also treated cultures of dissociated supporting cells for 5 days with 1 mM VPA; we observed no change in the numbers of HCS-1- or calretinin-labeled cells in those cultures (data not shown).

Treatment with VPA does not affect hair cell differentiation during regeneration. Organotypic cultures of the chick utricle were treated for 24 h with 1 mM streptomycin, rinsed and maintained for 4 days in drug-free medium, and then treated for 4 days with 1 mM VPA. Cultures were fixed and HCS-1-labeled hair cells were quantified. Treatment with VPA caused no significant change in hair cell recovery (p > 0.10), suggesting that HDAC inhibition does not affect hair cell differentiation. Error bars: standard deviation.
Discussion
Hair cells in the avian cochlea and vestibular organs are quickly regenerated after ototoxic injury (Corwin and Oberholtzer 1997). Replacement hair cells are generated by epithelial supporting cells, either through renewed proliferation or by direct phenotypic change (e.g., Raphael 1992; Roberson et al. 2004; Weisleder and Rubel 1993). Both of these regenerative processes will require significant changes in gene expression (e.g., Hawkins et al. 2007), but the possible involvement of epigenetic forms of regulation (e.g., histone acetylation) in regeneration has not been previously examined. In the present study, we show that pharmacological inhibition of histone deacetylases results in decreased proliferation of vestibular supporting cells, both in dissociated culture and in intact utricles. Nearly identical results were obtained using four different HDAC inhibitors from three distinct chemical classes, arguing that the reduction in proliferation was caused by changes in chromatin structure, rather than by indirect effects. We also found that treatment with the HDAC inhibitor valproic acid caused a reduction in the numbers of regenerated hair cells but did not appear to directly affect hair cell differentiation. This result is consistent with the notion that the primary effect of HDAC inhibition was a reduction in supporting cell proliferation.
Previous studies, which examined a variety of cell phenotypes, have shown that treatment with HDAC inhibitors leads to reduced levels of cell division (Bolden et al. 2006; Hsieh et al. 2004; Lu and Corwin 2008). The precise mechanisms that relate histone acetylation to cell cycle entry are not known, but a number of candidate signaling pathways have been identified. Expression of the cell cycle inhibitor p27kip, for example, is increased in response to treatment with HDAC inhibitors (Chen and Faller 2005; Fandy et al. 2005; Mayo et al. 2007; Platta et al. 2007; Sakajiri et al. 2005). In mammalian supporting cells, developmental expression of p27kip is correlated with cell cycle exit and genetic deletion of p27kip leads to ongoing proliferation (Chen and Segil 1999; Lowenheim et al. 1999; White et al. 2006). Notably, expression of p27kip is also downregulated during the onset of regenerative proliferation in the chick utricle (Hawkins et al. 2007; White et al. 2006). Additional studies have shown that HDAC inhibition leads to increased expression of the cell cycle inhibitors p19 (Ink4d) and p21 (CIP1/WAF1; Ju and Muller 2003; Okamoto et al. 2006; Richon et al. 2000; Rocchi et al. 2005; Yokota et al. 2004), and these molecules have also been implicated in the maintenance of mammalian supporting cells in a postmitotic state (Laine et al. 2007). Finally, expression of cyclins D and E—which are activators of proliferation—is decreased in both mammalian and avian cells following treatment with HDAC inhibitors (Finzer et al. 2001; Kim et al. 2007a, b; Nemajerova et al. 2003; Sandor et al. 2000).
Histone acetylation is most often associated with the “closing” of nuclear chromatin, leading to an inhibition of transcription and decreased gene expression (e.g., Kondo 2006). Conversely, preventing histone deacetylation (via treatment with HDAC inhibitors) should allow maintained expression of certain target genes. Our observation that treatment with HDAC inhibitors leads to a profound reduction in regenerative proliferation suggests that quiescent supporting cells may be expressing genes that inhibit cell cycle entry. It would be of great interest to identify genes that actively inhibit the proliferation of supporting cells since they may partially account for the qualitative differences in regenerative ability of the avian vs. mammalian ear. Several previous studies have utilized microarray methods to characterize gene expression in the regenerating avian ear (Hawkins et al. 2003, 2007), and further studies should reveal the identity of genes in the avian ear whose expression is regulated by histone acetylation.
Inhibition of HDAC1, 2, and 3 is sufficient for decreased proliferation
Using HDAC inhibitors with different class specificities, we found that inhibition of HDAC1, 2, and 3 is sufficient to reduce the proliferation of supporting cells. Specifically, we compared the effects of the inhibitor MS-275 (which strongly inhibits certain class I HDACs but has no appreciable activity against other HDAC classes) with TSA (Hu et al. 2003; Khan et al. 2008). We found that treatments with either MS-275 or TSA resulted in comparable decreases in proliferation and that their effects were not additive. We also used PCR techniques to demonstrate the presence of both HDAC1 and HDAC2 (type I HDACs that are inhibited by MS-275) within the sensory epithelium of the utricle. The structures of HDAC1 and HDAC2 are highly conserved, although their functions are not completely redundant (Gregoretti et al. 2004). HDAC1 and HDAC2 are contained within cell nuclei and interact with many protein complexes that have diverse functions (Grozinger and Schreiber 2002; Takami et al. 1999). In contrast, expression of class II HDACs is tissue specific, with higher levels present in brain and skeletal muscle (Gray and Ekstrom 2001). The function of class II HDACs has not been completely characterized. Rather than being confined to cell nuclei, they appear to shuttle between the nucleus and cytoplasm (Lemercier et al. 2000; Miska et al. 1999). Class II HDACs also appear to interact with different signaling complexes than do class I HDACs (Verdin et al. 2003). These observations suggest that class I and class II HDACs may serve fundamentally different roles in cellular signaling.
Effects of HDAC inhibition on hair cell differentiation
Treatment with VPA during the final phase of the regenerative process (i.e., recovery days 5–8) did not reduce the number of replacement hair cells. Since this is the time period during which many newly produced cells undergo differentiation, the result suggests that HDAC activation is not required for normal acquisition of the hair cell phenotype (Matsui et al. 2000; Stone and Rubel 2000). On the other hand, HDAC inhibition for the entire recovery period resulted in ∼50% reduction in the numbers of replacement hair cells (Fig. 5). While this was a significant reduction, it is still smaller than would be expected if replacement hair cells were produced by renewed proliferation alone. Treatment with VPA during the early (proliferative) phase of regeneration resulted in a ∼70% decrease in supporting cell proliferation. Thus, if all replacement hair cells were created by cell division, we would have expected a comparable decrease in hair cell recovery. This discrepancy could be explained if we assume that some replacement hair cells were produced through direct transdifferentiation. Studies of the amphibian ear have suggested that, under mitotically blocked conditions, new hair cells still arise through a direct change in cell phenotype (e.g., Baird et al. 2000; Taylor and Forge 2005). In addition, about 30% of the recovery observed in the regenerating chick cochlea has been attributed to direct transdifferentiation (Roberson et al. 2004).
Treatment with HDAC inhibitors does not reduce streptomycin ototoxicity
The observed discrepancy between regenerative proliferation and recovered hair cell numbers might also suggest a VPA-induced otoprotection of hair cells. A previous report has indicated that in vivo treatment with NaB can partially prevent cisplatin ototoxicity (Drottar et al. 2006). In order to examine this possibility, we compared hair cell survival in utricles that were cotreated with VPA and streptomycin to those treated with streptomycin alone. Notably, we did not observe any change in hair cell numbers, and we conclude that culture with VPA does not confer otoprotection from streptomycin toxicity. This result further suggests that direct transdifferentiation of supporting cells into hair cells accounts for the larger than expected level of hair cell recovery that was observed after 8 days of VPA treatment.
Apoptosis caused by higher concentration of HDAC inhibitors
Finally, we observed that treatment with HDAC inhibitors could also lead to increased apoptosis. It is notable that these cytotoxic effects were observed mostly when dissociated supporting cells were treated with high concentrations of HDAC inhibitors (i.e., doses that were higher than those required to reduce proliferation). The basis for the apoptotic effect is not known, but it could be due to off-target activities of these compounds. In contrast to the other inhibitors, treatment with VPA caused minimal toxicity. Similarly, low levels of apoptosis have been reported in cultured neural stem cells, following treatment with comparable doses of VPA (Hsieh et al. 2004).
In summary, our results suggest an important role for HDAC activation in cell cycle regulation of avian supporting cells. Since treatment with HDAC inhibitors causes changes in chromatin structure, it is likely that the effects of HDAC inhibitors are due to resulting changes in gene expression. It would be of great interest to identify the specific genes that are targeted by HDAC inhibition, which should help to elucidate the role these individual genes play in the regenerating chick ear. Identification of specific genes that regulate cell cycle entry in the mature ear should also aid in the development of mechanisms to initiate regeneration in the human inner ear.
References
Baird RA, Burton MD, Fashena DS, Naeger RA. Hair cell recovery in mitotically blocked cultures of the bullfrog saccule. Proc. Natl. Acad. Sci. U. S. A. 97:11722–11729, 2000.
Balasubramaniyan V, Boddeke E, Bakels R, Kust B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 143:939–951, 2006.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5:769–784, 2006.
Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr. Opin. Pharmacol. 6:369–375, 2006.
Chen JS, Faller DV. Histone deacetylase inhibition-mediated post-translational elevation of p27KIP1 protein levels is required for G1 arrest in fibroblasts. J. Cell Physiol. 202:87–99, 2005.
Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 126:1581–1590, 1999.
Corwin JT, Oberholtzer JC. Fish n’ chicks: model recipes for hair-cell regeneration? Neuron. 19:951–954, 1997.
Cyr JL, Warchol ME, Richardson GP, Corwin JT. Identification of the hair cell soma antigen HCS-1 as otoferlin. Assoc. Res. Otolaryngol. Abstr. 665, 2006.
Drottar M, Liberman MC, Ratan RR, Roberson DW. The histone deacetylase inhibitor sodium butyrate protects against cisplatin-induced hearing loss in guinea pigs. Laryngoscope. 116:292–296, 2006.
Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia. 7:646–657, 2005.
Finzer P, Kuntzen C, Soto U, zur Hausen H, Rosl F. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene. 20:4768–4776, 2001.
Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20:6969–6978, 2001.
Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp. Cell Res. 262:75–83, 2001.
Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338:17–31, 2004.
Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9:3–16, 2002.
Hawkins RD, Bashiardes S, Helms CA, Hu L, Saccone NL, Warchol ME, Lovett M. Gene expression differences in quiescent versus regenerating hair cells of avian sensory epithelia: implications for human hearing and balance disorders. Hum. Mol. Genet. 12:1261–1272, 2003.
Hawkins RD, Bashiardes S, Powder KE, Sajan SA, Bhonagiri V, Alvarado DM, Speck J, Warchol ME, Lovett M. Large scale gene expression profiles of regenerating inner ear sensory epithelia. PLoS ONE. 2:e525, 2007.
Hsieh J, Gage FH. Epigenetic control of neural stem cell fate. Curr. Opin. Genet. Dev. 14:461–469, 2004.
Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 101:16659–16664, 2004.
Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 307:720–728, 2003.
Jenuwein T, Allis CD. Translating the histone code. Science. 293:1074–1080, 2001.
Ju R, Muller MT. Histone deacetylase inhibitors activate p21(WAF1) expression via ATM. Cancer Res 63:2891–2897, 2003.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26:239–257, 1972.
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409:581–589, 2008.
Kim J, Park H, Im JY, Choi WS, Kim HS. Sodium butyrate regulates androgen receptor expression and cell cycle arrest in human prostate cancer cells. Anticancer. Res. 27:3285–3292, 2007a.
Kim YH, Lim JH, Lee TJ, Park JW, Kwon TK. Expression of cyclin D3 through Sp1 sites by histone deacetylase inhibitors is mediated with protein kinase C-delta (PKC-delta) signal pathway. J. Cell Biochem. 101:987–995, 2007b.
Kondo T. Epigenetic alchemy for cell fate conversion. Curr. Opin. Genet. Dev. 16:502–507, 2006.
Kouzarides T. Chromatin modifications and their function. Cell. 128:693–705, 2007.
Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell. Biochem. 42:65–82, 1982.
Laine H, Doetzlhofer A, Mantela J, Ylikoski J, Laiho M, Roussel MF, Segil N, Pirvola U. p19(Ink4d) and p21(Cip1) collaborate to maintain the postmitotic state of auditory hair cells, their codeletion leading to DNA damage and p53-mediated apoptosis. J. Neurosci. 27:1434–1444, 2007.
Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J. Biol. Chem. 275:15594–15599, 2000.
Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 128:707–719, 2007.
Lowenheim H, Furness DN, Kil J, Zinn C, Gultig K, Fero ML, Frost D, Gummer AW, Roberts JM, Rubel EW, Hackney CM, Zenner HP. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of Corti. Proc. Natl. Acad. Sci. U. S. A. 96:4084–4088, 1999.
Lu Z, Corwin JT. The influence of glycogen synthase kinase 3 in limiting cell addition in the mammalian ear. Dev. Neurobiol. 68:1059–1075, 2008.
Matsui JI, Oesterle EC, Stone JS, Rubel EW. Characterization of damage and regeneration in cultured avian utricles. J. Assoc. Res. Otolaryngol. 1:46–63, 2000.
Matsui JI, Ogilvie JM, Warchol ME. Inhibition of caspases prevents ototoxic and ongoing hair cell death. J. Neurosci. 22:1218–1227, 2002.
Mayo C, Lloreta J, Real FX, Mayol X. In vitro differentiation of HT-29 M6 mucus-secreting colon cancer cells involves a trichostatin A and p27(KIP1)-inducible transcriptional program of gene expression. J. Cell Physiol. 212:42–50, 2007.
Michaelis M, Michaelis UR, Fleming I, Suhan T, Cinatl J, Blaheta RA, Hoffmann K, Kotchetkov R, Busse R, Nau H, Cinatl J, Jr. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol. Pharmacol. 65:520–527, 2004.
Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18:5099–5107, 1999.
Nemajerova A, Smarda J, Jurdic P, Kubala L, Soucek K, Smardova J. Trichostatin A suppresses transformation by the v-myb oncogene in BM2 cells. J. Hematother. Stem Cell Res. 12:225–235, 2003.
Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk. Res. 32:1382–1392, 2008.
Okamoto H, Fujioka Y, Takahashi A, Takahashi T, Taniguchi T, Ishikawa Y, Yokoyama M. Trichostatin A, an inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21(WAF1). J. Atheroscler. Thromb. 13:183–191, 2006.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276:36734–36741, 2001.
Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. The HDAC inhibitor trichostatin A inhibits growth of small cell lung cancer cells. J. Surg. Res. 142:219–226, 2007.
Raphael Y. Evidence for supporting cell mitosis in response to acoustic trauma in the avian inner ear. J. Neurocytol. 21:663–671, 1992.
Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U. S. A. 97:10014–10019, 2000.
Roberson DW, Alosi JA, Cotanche DA. Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J. Neurosci. Res. 78:461–471, 2004.
Rocchi P, Tonelli R, Camerin C, Purgato S, Fronza R, Bianucci F, Guerra F, Pession A, Ferreri AM. p21Waf1/Cip1 is a common target induced by short-chain fatty acid HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in neuroblastoma cells. Oncol. Rep. 13:1139–1144, 2005.
Rogers JH. Immunoreactivity for calretinin and other calcium-binding proteins in cerebellum. Neuroscience. 31:711–721, 1989.
Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. U. S. A. 96:4592–4597, 1999.
Sajan SA, Warchol ME, Lovett M. Towards a systems biology of mouse inner ear organogenesis: gene expression pathways, patterns and network analysis. Genetics 177:631–663, 2007.
Sakajiri S, Kumagai T, Kawamata N, Saitoh T, Said JW, Koeffler HP. Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines. Exp. Hematol. 33:53–61, 2005.
Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer. 83:817–825, 2000.
Stone JS, Rubel EW. Cellular studies of auditory hair cell regeneration in birds. Proc. Natl. Acad. Sci. U. S. A. 97:11714–11721, 2000.
Takami Y, Kikuchi H, Nakayama T. Chicken histone deacetylase-2 controls the amount of the IgM H-chain at the steps of both transcription of its gene and alternative processing of its pre-mRNA in the DT40 cell line. J. Biol. Chem. 274:23977–23990, 1999.
Taylor RR, Forge A. Hair cell regeneration in sensory epithelia from the inner ear of a urodele amphibian. J. Comp. Neurol. 484: 105–120, 2005.
Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends. Genet. 19:286–293, 2003.
Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs. 13:1–13, 2002.
Warchol ME. Supporting cells in isolated sensory epithelia of avian utricles proliferate in serum-free culture. Neuroreport. 6:981–984, 1995.
Warchol ME. Immune cytokines and dexamethasone influence sensory regeneration in the avian vestibular periphery. J. Neurocytol. 28:889–900, 1999.
Warchol ME. Lectin from Griffonia simplicifolia identifies an immature-appearing subpopulation of sensory hair cells in the avian utricle. J. Neurocytol. 30:253–264, 2001.
Warchol ME. Cell density and N-cadherin interactions regulate cell proliferation in the sensory epithelia of the inner ear. J. Neurosci. 22:2607–2616, 2002.
Weisleder P, Rubel EW. Hair cell regeneration after streptomycin toxicity in the avian vestibular epithelium. J. Comp. Neurol. 331:97–110, 1993.
White PM, Doetzlhofer A, Lee YS, Groves AK, Segil N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 441:984–987, 2006.
Yokota T, Matsuzaki Y, Miyazawa K, Zindy F, Roussel MF, Sakai T. Histone deacetylase inhibitors activate INK4d gene through Sp1 site in its promoter. Oncogene. 23:5340–5349, 2004.
Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265:17174–17179, 1990.
Acknowledgements
This work is supported by grants R01 DC006283, T32 DC000022, and P30 DC04665 from the NIDCD (National Institutes of Health). We thank Dr. Kevin Ohlemiller for assistance with statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Slattery, E.L., Speck, J.D. & Warchol, M.E. Epigenetic Influences on Sensory Regeneration: Histone Deacetylases Regulate Supporting Cell Proliferation in the Avian Utricle. JARO 10, 341–353 (2009). https://doi.org/10.1007/s10162-009-0166-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-009-0166-y