
Abstract
Type 2 diabetes (T2D) has a complex etiology which is not yet fully elucidated. The identification of gene perturbations and hub genes of T2D may deepen our understanding of its genetic basis. We aimed to identify highly perturbed genes and hub genes associated with T2D via an extensive bioinformatics analytic workflow consisting of five steps: systematic review of Gene Expression Omnibus and associated literature; identification and classification of differentially expressed genes (DEGs); identification of highly perturbed genes via meta-analysis; identification of hub genes via network analysis; and downstream analysis of highly perturbed genes and hub genes. Three meta-analytic strategies, random effects model, vote-counting approach, and p value combining approach, were applied. Hub genes were defined as those nodes having above-average betweenness, closeness, and degree in the network. Downstream analyses included gene ontologies, Kyoto Encyclopedia of Genes and Genomes pathways, metabolomics, COVID-19-related gene sets, and Genotype-Tissue Expression profiles. Analysis of 27 eligible microarrays identified 6284 DEGs (4592 downregulated and 1692 upregulated) in four tissue types. Tissue-specific gene expression was significantly greater than tissue non-specific (shared) gene expression. Analyses revealed 79 highly perturbed genes and 28 hub genes. Downstream analyses identified enrichments of shared genes with certain other diabetes phenotypes; insulin synthesis and action-related pathways and metabolomics; mechanistic associations with apoptosis and immunity-related pathways; COVID-19-related gene sets; and cell types demonstrating over- and under-expression of marker genes of T2D. Our approach provided valuable insights on T2D pathogenesis and pathophysiological manifestations. Broader utility of this pipeline beyond T2D is envisaged.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to an analysis of global data through years 1990–2018, diabetes was prevalent in almost half a billion people, a number expected to rise by 25% and 51%, respectively, by 2030 and 2045 (Saeedi et al. 2019). Type 2 diabetes (T2D) accounts for over 90% of all diabetes cases (Zheng et al. 2018), affecting nearly 6.8% (537 million) of the world population in year 2021 (Sun et al. 2022). Current evidence suggests a complex and multifactorial etiology of T2D characterized by genetic and environmental interactions (Arroyo et al. 2021), although T2D pathogenesis is not yet fully elucidated. There are likely varying degrees of shared genetic origins in the pathogenesis of T2D and other diabetes phenotypes such as type 1 diabetes (T1D) (Aylward et al. 2018), latent autoimmune diabetes in adults (Basile et al. 2014), and maturity-onset diabetes of the young (MODY) (Bonnefond et al. 2020). Recent studies also support the deconstruction of T2D heterogeneity to define T2D sub-types (Udler et al. 2018) and the delineation of a continuum of diabetes sub-types (Flannick et al. 2016) instead of the status quo characterized by a few distinct diabetes phenotypes. Understanding the genetic basis of T2D is fundamental to precision medicine approaches striving for impeccable matching and an individualized level of T2D care (Prasad and Groop 2019).
Cardinal tissues of the body impacted by heightened insulin resistance and diminished insulin secretion in T2D include the pancreas, liver, skeletal muscle, and adipose tissue (Batista et al. 2021). Exploration of gene perturbations in these tissues can deepen our understanding of the molecular etio-pathology of T2D. Highly up- and downregulated genes expressed consistently across different tissue types may uncover potential genome-wide biomarkers or “gene signatures,” which are integral to achieving precision diagnostic, prognostic, monitoring, and treatment approaches. Topologically, hub genes are defined as highly and tightly connected nodes in typically scale-free gene regulatory networks (GRN) (Buchberger et al. 2021). As such, criteria such as high correlation in candidate modules (Liu et al. 2019a) and above-average betweenness, closeness, and degree (Liu et al. 2019b) in GRN have been used to demarcate hub genes in previous studies. Functionally, they perform critical regulatory roles in biological processes interacting with many other genes in associated pathways. Given their crucial structural and functional characteristics, hub genes are highly sought-after in precision medicine approaches as plausible niches for developing drug and treatment targets (Liu et al. 2021). In this context, the importance of identifying highly perturbed genes and hub genes associated with T2D for the purpose of individualizing T2D care is unequivocal.
Downstream analyses of gene sets provide invaluable insights into associated core biological functions, pathways, diseases, drugs, and many other aspects. Frequently used gene ontology (GO) analysis provides evidence as a snapshot of contemporary biological knowledge related to a given gene including its function at the molecular level, the cellular location(s) it functions at, and the biological processes reliant on it (Hill et al. 2008). Pathway analyses such as Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000) entail derivation of coherent and meaningful biological phenomena attributable to input genes (Nguyen et al. 2019). Additional downstream analyses of diseases, drugs, metabolomes, and tissue enrichment are also available and can provide valuable insights into associated genes. Taken together, downstream analysis of highly perturbed and hub genes associated with T2D may render valuable information on aspects such as affected biological processes, dysregulated pathways, related diseases, and metabolomic biomarkers.
Advances in high-throughput technologies have generated a wealth of gene expression data, while the availability of open-source platforms such as the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) (Edgar et al. 2002; Barrett et al. 2013) and simultaneous advent in big data and bioinformatics analytic tools such as microarray and RNA-seq meta-analysis and gene–gene interaction network analysis strategies have offered unprecedented opportunities for high-level evidence synthesis from a multitude of gene expression datasets. Such approaches are likely to render new knowledge on complex diseases like T2D and acquire adequate statistical power to identify genes associated with a disease that may not have been evoked via prior analysis of a single or a few datasets.
Yet, to date, no comprehensive evidence synthesis study has been performed to identify highly perturbed genes and hub genes associated with T2D in human adults using an extensive bioinformatics analytic pipeline. Prior studies were limited to identifying hub genes in a few (n = 3) microarrays from a single tissue type (pancreatic islets) (Lin et al. 2020) or performing an ad hoc gene expression meta-analysis of all diabetes phenotypes (n = 13) (Mei et al. 2017). In this study, we aimed to identify highly perturbed genes and hub genes associated with T2D in different tissues of adult humans via a pre-defined and extensive bioinformatics analytic workflow consisting of systematic review, meta-analysis, identification and classification of differentially expressed genes (DEGs), network analysis, and downstream analysis.
Methods
The methodological approach consisted of five sequential steps: (1) systematic review of NCBI GEO expression data and related publications, (2) analysis of microarrays to identify DEGs, (3) meta-analysis of DEGs to identify highly perturbed genes in T2D, (4) network analysis of gene–gene/protein–protein interactions to identify hub genes in T2D, and (5) downstream analysis of highly perturbed genes and hub genes associated with T2D.
Systematic review
A preliminary search on the NCBI GEO database was first run on 1st February 2021 using a pre-defined search string: “(“diabetes mellitus, type 2” [MeSH Terms] OR type 2 diabetes[All Fields]) AND “Homo sapiens” [porgn] AND “Expression profiling by array” [Filter].” The resulting microarrays and related publications were further screened against pre-defined eligibility criteria. Microarrays of conditions other than T2D, all diabetes phenotypes other than T2D, and early dysglycemic conditions such as impaired glucose tolerance, insulin resistance, and prediabetes were excluded. Studies with non-human specimens, children, without healthy controls, or with notable comorbidity in control samples were also excluded. We also omitted studies involving long non-coding RNA (lncRNA), micro RNA (miRNA), samples subject to drug treatments and other interventions, pluripotent stem cells, xenografts, transfected or transgenic tissues, undifferentiated tissues, and sub-samples in super-series. Microarrays passing these eligibility criteria were selected for manual curation which were then further screened along with the full texts of related publications (where available) for the presence of adequate information such as clinical diagnosis (healthy vs T2D) and gene symbol/Entrez ID. Following this, all microarrays with sufficient information were selected for subsequent analyses.
Identification of DEGs
All microarrays selected from the systematic review were imported to R using getGEO function of GEOquery package (Davis and Meltzer 2007). In each expression set, phenoData component was examined to determine the number of eligible samples and confirm the presence of outcome (T2D vs controls) variable, while featureData component was explored to verify the presence of gene annotation information. Where required, non-normalized gene expression matrices were log2 transformed in order to alleviate skewness and create symmetric distributions (Le et al. 2020). Samples were assessed for the presence of any batch effects between the two groups by running principal components analysis on transposed expression matrices and were rectified using removeBatchEffect function in limma package (Ritchie et al. 2015). Relevant features were annotated with expression matrices to generate curated data for running differential gene expression analysis. Samples were ascribed to the relevant group (T2D/control) using model.matrix function of limma package (Ritchie et al. 2015) producing a binary design matrix. As the detection of DEGs can be enhanced by filtering genes with a low expression level, we assumed a median (50%) cut-off for the gene expression level. A uniform analytic pipeline consisting of the following sequential steps was applied to each microarray: (1) median expression levels were calculated, and those above the median were retained. (2) From the resulting genes, those expressed in more than two samples were retained, while the others were removed. (3) Model fitting was performed using lmFit function of limma (Ritchie et al. 2015) to enumerate expression levels of T2D and control groups. (4) Contrasts were defined as “T2D, control,” and empirical Bayes step was run to derive differential expression results. (5) The DEGs, defined as those with log2FC > 1 and Benjamini–Hochberg (BH)-adjusted p < 0.05 for upregulated genes and log2FC < − 0.5 and BH-adjusted p < 0.05 for downregulated genes, were identified for each microarray.
Microarrays with no DEGs were excluded, and those with non-zero DEGs were visualized with a clustered bar chart. Furthermore, DEGs were classified by tissue types and visualized as a Venn diagram. Information on clinical and other features of the microarrays with non-zero DEGs, individual DEGs identified by each dataset, and tissue-based classification of DEGs were also summarized.
Meta-analysis of DEGs: identification of highly perturbed genes
In order to identify highly perturbed genes associated with T2D, we conducted meta-analysis of DEGs using MetaVolcanoR package (Prada et al. 2020). We implemented all 3 meta-analytic strategies incorporated in this package, namely, random effects model (REM), vote-counting approach (VC), and p value combining approach (CA).
In brief, REM synthesizes a summary fold-change of multiple microarrays based on variance, producing a summary p value which indicates the probability that the summary fold-change is not different to zero. The metathr parameter can be specified to filter the desired percentage of the top-most consistently perturbed genes. Gene perturbation is ranked as per the topconfects approach (Harrison et al. 2019). The VC algorithm produces highly perturbed genes according to user-specified p values and fold-change cut-off levels, taking into account both the number of studies in which a DEG appeared and its gene fold-change sign consistency. Here also, metathr parameter can be defined to extract the required percentage of highly perturbed genes. Meta-synthesis of gene perturbation by CA algorithm is at the mean or median level along with p values derived by Fisher method. A required proportion of top-most DEGs can be identified by specifying metathr parameter with CA as well (Prada et al. 2020).
As required by the package, all microarray datasets with non-zero DEGs; each consisting of the columns gene name (symbol), fold-change (log2FC), and p value; and confidence intervals of the fold-change (CI.L and CI.R) were merged to build a list item. For all 3 meta-analytic models, metathr was set at 0.01. For VC, p value and absolute fold-change thresholds were set at 0.05 and 0, respectively.
Highly perturbed genes identified by each model as well as the compiled list of all highly perturbed genes were presented in tabular format. Volcano plots were drawn to illustrate the top-most perturbed genes identified by REM, VC, and CA methods. Inverse cumulative distribution of consistently differentially expressed genes as per VC was plotted to demonstrate the number of genes with perturbed expression in ≥ 1 studies. Detailed meta-analytic outputs from all 3 approaches and the highly perturbed genes identified by each method were also compiled.
Network analysis: identification of hub genes
The list of highly perturbed genes was fed into GENEMANIA (Warde-Farley et al. 2010) to determine the gene–gene interaction network. The interaction network formulated by the GENEMANIA gene function prediction program, based on the multiple association network integration algorithm (MANIA), incorporates a multitude of functional associations including co-expression, pathways, physical interactions, co-localization, genetic interactions, and protein domain similarity. It has been found more accurate and computationally efficient than other gene function prediction methods (Mostafavi et al. 2008; Peña-Castillo et al. 2008). The gene–gene interaction network was first constructed and visualized in GENEMANIA. Next, these interactions were imported to visualize the protein–protein interaction (PPI) network in STRING version 11.0 (Szklarczyk et al. 2019). We used the Centiscape application (Scardoni et al. 2009) in the Cytoscape software (Shannon et al. 2003) to analyze the PPI network and determine the hub nodes. After removing nodes with a connection number < 2, the network was visualized in Cytoscape. Hub genes were defined as those nodes in the network with betweenness, closeness, and degree higher than their mean values. A similar approach has been previously used to demarcate hub genes (Liu et al. 2019b). Topological features of the hub nodes and details of the PPI network derived by Centiscape were summarized.
Downstream analysis of highly perturbed genes and hub genes
Using Enrichr (Chen et al. 2013; Kuleshov et al. 2016; Xie et al. 2021) platform, we ran a series of downstream analyses for both highly perturbed genes and hub genes as outlined below:
-
Ontologies: GO Biological Process 2018 (Hill et al. 2008)
-
Pathways: KEGG 2019 Human (Kanehisa and Goto 2000)
-
Diseases/drugs: COVID-19-related gene sets
-
Cell types: GTEx tissue sample gene expression profiles up and GTEx tissue sample gene expression profiles down
-
Miscellaneous: HMDB metabolites
The Genotype-Tissue Expression (GTEx) portal contains tissue-specific gene expression and regulation data (GTEx Consortium 2013), whereas the Human Metabolome Database (HMDB) records human metabolomics data (Wishart et al. 2018).
Results
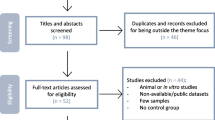
The bioinformatic analytic workflow is summarized in Online Resource 1.
Systematic review outputs
The preliminary search resulted in 178 eligible microarrays, while 45 of these were selected for manual curation. We selected 27 microarrays with sufficient information for subsequent analyses, the details of which are provided in Online Resource 2.
Differential gene expression analysis outputs
The number of DEGs identified by each dataset is shown in Table 1. There were 11 microarrays with no DEGs. In Fig. 1, microarrays with non-zero DEGs (n = 16) are visualized as a clustered bar chart. The identified DEGs belonged to four tissue types (i.e., circulatory; adipose; digestive; skeletal muscle) as visualized in the Venn diagram (Fig. 2). Clinical and other information of the 16 microarrays with non-zero DEGs is presented in Online Resource 3. Details of DEGs identified by each dataset are presented in Online Resource 4, while DEGs classified by tissue type are presented in Online Resource 5.

Clustered bar chart depicting the number of differentially expressed (up- and downregulated) genes in the 16 datasets

Venn diagram depicting the number of differentially expressed genes by the 4 tissue types
A significantly larger proportion of genes associated with T2D is downregulated
Of all the DEGs identified by different tissues (n = 6284), the proportion of downregulated genes (n = 4592) was significantly higher than the proportion of upregulated genes (n = 1692) (p < 0.00000001). At the level of tissue type, circulatory (255 downregulated vs 94 upregulated; p < 0.000001), adipose (1426 downregulated vs 1212 upregulated; p = 0.000017), and digestive (2664 downregulated vs 135 upregulated; p < 0.000001) tissues revealed a similar pattern, while the skeletal muscle (247 downregulated vs 251 upregulated; p = 0.44653) showed no such over-expression of downregulated genes. Of the 27 microarrays, 11 contained no DEGs, while the two datasets containing the highest number of DEGs were from pancreatic islets (GSE25724; 2164 DEGs) and visceral adipose tissue (GSE29231; 1655 DEGs) (Table 1, Fig. 1).
Tissue-specific DEGs are predominant compared to DEGs shared between tissues in T2D
Proportions of tissue-specific DEGs were significantly higher than the proportions shared with one or more other tissue types in circulatory (242 specific vs 89 non-specific; p < 0.000001), adipose (1989 specific vs 342 non-specific; p < 0.000001), and digestive (1875 specific vs 409 non-specific; p < 0.000001) groups, while skeletal muscle (244 specific vs 217 non-specific; p = 0.112959) had no such over-expression (Fig. 2).
Meta-analytic outputs
As shown in Table 2, the three meta-analytic algorithms REM, VC, and CA identified 49, 27, and 8 highly perturbed genes, respectively. The compiled list, after removing redundancies, comprised 79 highly perturbed genes. Volcano plots illustrating the top-most perturbed genes identified by REM, VC, and CA are given in Figs. 3, 4, 5, respectively. Figure 6 presents the inverse cumulative distribution of consistently differentially expressed genes as per VC, plotted to demonstrate the number of genes with perturbed expression in ≥ 1 studies. We present detailed meta-analytic outputs from all 3 approaches in Online Resource 6 and highly perturbed genes identified by each method in Online Resource 7.

Highly perturbed genes (n = 49) identified by random effects model meta-analysis in MetaVolcanoR package with metathr set at 0.01. Consistently upregulated genes appear in red and consistently downregulated genes appear in blue

Highly perturbed genes (n = 27) identified by vote-counting approach meta-analysis in MetaVolcanoR package with metathr set at 0.01. Consistently upregulated genes appear in red and consistently downregulated genes appear in blue

Highly perturbed genes (n = 8) identified by p value combining approach meta-analysis in MetaVolcanoR package with metathr set at 0.01. Consistently upregulated genes appear in red and consistently downregulated genes appear in blue

Inverse cumulative distribution of the consistently differentially expressed genes as per vote-counting approach
Highly perturbed genes associated with T2D comprise both up- and downregulated genes
There was no significant difference (p = 0.410983) between the proportions of up- (38/79) and downregulated (41/79) genes that constituted the highly perturbed gene set (n = 79) identified by all 3 meta-analytic algorithms (Table 2, Figs. 3, 4, 5, 6). The 38 upregulated genes were ALDOB; BCL3; CNTFR; CNTNAP2; CRTAC1; DYRK3; EGR2; ELFN1; ERAP2; ESPNL; ISLR; LOC100008589; LOC644422; LOC649456; MCL1; MGRN1; MMP9; NPTX2; PCOLCE2; PHACTR3; PHLDA1; PI3; POMZP3; PRIMA1; PVRL2; RASL11B; RNF19B; SCN1B; SLC9A3R2; SOD3; TAPBP; TNFAIP6; U2AF2; VNN2; XYLT1; ZBTB16; ZNF423; and ZNF75. The 41 downregulated genes were API5; APOL4; ARG2; ASCL2; C14orf132; C19orf33; COG2; CTSC; DHRS2; DYRK2; ENPP2; ENTPD3; FUT11; HADH; HLA-DRB4; HLA-DRB5; IAPP; IFNA7; KIAA1279; KIAA1984; LARP4; LOC389286; LOC650885; LOC731682; MARK1; MCOLN3; MTRR; NAALAD2; NMNAT2; OR8B12; PAAF1; POPDC3; PPM1K; PPP1R1A; RPL14; SLC2A2; SNAP25; STMN2; TAP2; TMEM37; and UGT2B7.
Network analysis outputs
Network analysis identified 28 hub genes, the topological features of which are summarized in Table 3. Figure 7 presents the gene–gene interaction network produced on GENEMANIA, and details of this network are given in Online Resource 8. The PPI network visualized on STRING version 11.0 (Szklarczyk et al. 2019) is given in Fig. 8. The network created by Cytoscape is provided in Fig. 9. Details of the PPI network derived by Centiscape are provided in Online Resource 9.

Gene–gene interactions network visualized in GENEMANIA

Protein–protein interactions network visualized in STRING

Two different presentations of the protein–protein interactions network in Centiscape
Hub genes associated with T2D also comprise both up- and downregulated genes
The 28 hub genes consisted of 13 upregulated, 9 downregulated, and 6 predicted genes. There was no significant difference (p = 0.261216) between the proportions of up- (13/22) and downregulated (9/22) genes constituting the hub gene set. The 13 upregulated hub genes were CNTFR; CNTNAP2; ISLR; MCL1; MMP9; PHLDA1; RASL11B; SCN1B; SOD3; TNFAIP6; XYLT1; ZBTB16; and ZNF423. The 9 downregulated genes were CTSC; ENPP2; HADH; PPP1R1A; RPL14; SLC2A2; SNAP25; STMN2; and UGT2B7. The 6 hub genes predicted by GENEMANIA were ARG1; CD226; DPEP1; PPP1R15A; TAP1; and ZMIZ1 (Table 3).
Downstream functional analysis outputs
Findings from downstream analyses of highly perturbed genes are summarized in Tables 4, 5, 6 and illustrated in Figs. 10, 11, 12 with details in Online Resource 10. Results from downstream analyses of hub genes are presented in Tables 7, 8, 9 and visualized in Figs. 13, 14, 15 with details in Online Resource 11.

Downstream analyses of highly perturbed genes (n = 79) associated with type 2 diabetes in different tissues of human adults: a GO biological processes, b KEGG pathways

Downstream analyses of highly perturbed genes (n = 79) associated with type 2 diabetes in different tissues of human adults: a COVID-19 related gene sets, b HMDB metabolites

Downstream analyses of highly perturbed genes (n = 79) associated with type 2 diabetes in different tissues of human adults: a GTEx tissue sample gene expression profiles down, b GTEx tissue sample gene expression profiles up

Downstream analyses of hub genes (n = 28) associated with type 2 diabetes in different tissues of human adults: a GO biological processes, b KEGG pathways

Downstream analyses of hub genes (n = 28) associated with type 2 diabetes in different tissues of human adults: a COVID-19-related gene sets, b HMDB metabolites

Downstream analyses of hub genes (n = 28) associated with type 2 diabetes in different tissues of human adults: a GTEx tissue sample gene expression profiles down, b GTEx tissue sample gene expression profiles up
Enriched biological processes associated with highly perturbed genes and hub genes of T2D
Ontology analysis of highly perturbed genes identified 89 enriched biological processes (Online Resource 10). The top 10 enriched biological processes were intrinsic apoptotic signaling pathway in response to DNA damage (GO:0,008,630); intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator (GO:0,042,771); antigen processing and presentation of peptide antigen via MHC class I (GO:0,002,474); cellular response to oxidative stress (GO:0,034,599); plasma membrane bounded cell projection organization (GO:0,120,036); intrinsic apoptotic signaling pathway by p53 class mediator (GO:0,072,332); myeloid cell differentiation (GO:0,030,099); nicotinamide nucleotide metabolic process (GO:0,046,496); regulation of intrinsic apoptotic signaling pathway (GO:2,001,242); and myeloid leukocyte differentiation (GO:0,002,573). Three of these, antigen processing and presentation of peptide antigen via MHC class I (GO:0,002,474); myeloid cell differentiation (GO:0,030,099), and regulation of intrinsic apoptotic signaling pathway (GO:2,001,242), were associated with upregulated genes in T2D (Table 4).
Ontological analysis of hub genes identified 179 enriched biological processes (Online Resource 11). Top 10 enriched biological processes were neutrophil degranulation (GO:0,043,312); neutrophil activation involved in immune response (GO:0,002,283); neutrophil-mediated immunity (GO:0,002,446); regulation of intrinsic apoptotic signaling pathway (GO:2,001,242); negative regulation of intrinsic apoptotic signaling pathway (GO:2,001,243); positive regulation of cell projection organization (GO:0,031,346); cellular response to reactive oxygen species (GO:0,034,614); negative regulation of apoptotic signaling pathway (GO:2,001,234); negative regulation of cysteine-type endopeptidase activity involved in apoptotic process (GO:0,043,154); and regulation of peptide hormone secretion (GO:0,090,276). Of these, three (regulation of intrinsic apoptotic signaling pathway (GO:2,001,242); negative regulation of intrinsic apoptotic signaling pathway (GO:2,001,243); negative regulation of apoptotic signaling pathway (GO:2,001,234)) were associated with upregulated genes in T2D, whereas one (regulation of peptide hormone secretion (GO:0,090,276)) was associated with downregulated genes in T2D (Table 6).
Enriched KEGG pathways associated with highly perturbed genes and hub genes of T2D
As per KEGG analysis of highly perturbed genes, 21 pathways were enriched (Online Resource 10). The top 10 pathways were Epstein–Barr virus infection; antigen processing and presentation; autoimmune thyroid disease; maturity-onset diabetes of the young; asthma; allograft rejection; graft-versus-host disease; type 1 diabetes mellitus; intestinal immune network for IgA production; and cell adhesion molecules (CAMs). Seven of these (autoimmune thyroid disease; maturity-onset diabetes of the young; asthma; allograft rejection; graft-versus-host disease; type 1 diabetes mellitus; intestinal immune network for IgA production) were associated with downregulated genes in T2D (Table 4).
According to KEGG analysis of hub genes, 12 pathways were enriched (Online Resource 11). The top 10 pathways were insulin secretion; apoptosis; adrenergic signaling in cardiomyocytes; cell adhesion molecules (CAMs); JAK-STAT signaling pathway; transcriptional mis-regulation in cancer; arginine biosynthesis; maturity-onset diabetes of the young; ascorbate and aldarate metabolism; and fatty acid elongation. Four of these (insulin secretion; maturity-onset diabetes of the young; ascorbate and aldarate metabolism; fatty acid elongation) were associated with downregulated genes in T2D, while two pathways (JAK-STAT signaling pathway and transcriptional mis-regulation in cancer) were associated with upregulated genes in T2D (Table 6).
COVID-19 related gene sets associated with highly perturbed genes and hub genes of T2D
Downstream analyses revealed 20 COVID-19-related gene sets associated with the highly perturbed genes of T2D (Online Resource 10), and the top 10 of these are visualized in Fig. 11a. There were 23 COVID-19-related gene sets associated with the hub genes of T2D (Online Resource 11), the top 10 of which are visualized in Fig. 14a.
HMDB metabolites associated with highly perturbed genes and hub genes of T2D
Four HMDB metabolites (zinc (HMDB01303), manganese (HMDB01333), magnesium (HMDB00547), C10H13N2O7P (HMDB01570)) were associated with the highly perturbed genes of T2D (Online Resource 10). Of these, Zinc (HMDB01303) was associated with upregulated genes in T2D, while C10H13N2O7P (HMDB01570) was associated with downregulated genes in T2D (Table 5).
There were 45 HMDB metabolites associated with the hub genes of T2D (Online Resource 11). The top 10 metabolites were zinc (HMDB01303); ethyl glucuronide (HMDB10325); 3-acetoacetyl-CoA (HMDB01484); (S)-methylmalonate semialdehyde (HMDB02217); C18H31NO14S (HMDB00632); ornithine (HMDB00214); 17beta-estradiol glucuronide (HMDB10317); (3alpha,5beta,20S)-20-hydroxypregnan-3-yl beta-D-glucopyranosiduronic acid (HMDB10318); 3,17-androstanediol glucuronide (HMDB10321); and 17alpha-estradiol-3-glucuronide (HMDB10322). Seven of these (Ethyl glucuronide (HMDB10325); 3-acetoacetyl-CoA (HMDB01484); (S)-methylmalonate semialdehyde (HMDB02217); 17beta-estradiol glucuronide (HMDB10317); (3alpha,5beta,20S)-20-hydroxypregnan-3-yl beta-D-glucopyranosiduronic acid (HMDB10318); 3,17-androstanediol glucuronide (HMDB10321); and 17alpha-estradiol-3-glucuronide (HMDB10322)) were associated with downregulated genes in T2D. One metabolite (C18H31NO14S (HMDB00632)) was associated with upregulated genes of T2D (Table 8).
Gene expression in different cell types associated with highly perturbed genes and hub genes of T2D
There were 168 downregulated GTEx profiles (Online Resource 10) associated with highly perturbed genes of T2D, the top 10 of which consisted of one brain, two thyroids, and seven blood tissue samples (Table 6 and Fig. 12). There were 77 upregulated GTEx profiles (Online Resource 10) associated with highly perturbed genes of T2D, the top 10 of which consisted of six adipose and one each of lung, breast, heart, and blood vessel tissue samples (Table 6 and Fig. 12).
There were 223 downregulated GTEx profiles (Online Resource 11) associated with hub genes of T2D, the top 10 of which consisted of five nervous system (three brain, one nerve, one pituitary), three blood, one breast, and one bladder tissue samples (Table 9 and Fig. 15). There were 178 upregulated GTEx profiles (Online Resource 11) associated with hub genes of T2D, the top 10 of which consisted of six adipose, two blood, and two skin tissue samples (Table 9 and Fig. 15).
Discussion
In this study, we identified highly perturbed genes and hub genes associated with T2D in different tissues of adult humans, via an extensive bioinformatics analytic workflow. Downstream analyses revealed valuable insights on T2D pathogenesis, including associations with other diabetes phenotypes and COVID-19 and patterns of tissue-specific and tissue non-specific differential gene expression as well as pathophysiological manifestations such as those related to insulin action, immunity, and apoptosis. Salient findings of the study which contribute towards the understanding of the genetic basis of T2D are further discussed below. The comprehensive evidence synthesis approach with open-source gene expression data exemplified in this study could be replicated to gain high-level evidence synthesis for other clinical conditions.
Patterns of differential gene expression in T2D
Our findings indicate that T2D seems rather a disorder of gene downregulation than upregulation, when the whole genome is considered. This is consistent with previous studies where a preponderance of gene downregulation was associated with T2D (Takematsu et al. 2020; Palsgaard et al. 2009). Also, hyperglycemia-induced global downregulation of gene expression in adipose and skeletal muscle tissues have been documented previously (Meugnier et al. 2007). A similar pattern has been observed in T1D (Yip et al. 2020) and other endocrine disorders such as polycystic ovary syndrome (Idicula-Thomas et al. 2020). In contrast, highly perturbed genes and hub genes associated with T2D, which might together constitute the candidate gene set critical for pathogenicity of T2D, were found to contain both up- and downregulated genes. This presentation suggests a more complex dysregulation at the crux of the GRN of T2D, involving actions and interactions between both repressed and augmented genes.
Tissue-specific and tissue non-specific DEGs associated with T2D
Results of the present study indicate the predominance of tissue-specific DEGs in T2D. This supports the use of target tissue gene expression analysis as a viable avenue for identifying tissue-specific T2D biomarkers. A previous analysis integrating multiple tissue transcriptomics and PPI data to explore molecular biomarkers of T2D confirmed the presence of common signatures (Calimlioglu et al. 2015). We also observed common DEGs across different tissue types which can act as confluent molecular signatures of T2D. Identification of tissue-specific and non-specific molecular gene signatures of T2D facilitates downstream exploration of key pathways amenable to therapeutic targeting and drug repurposing efforts.
Shared gene enrichment across diabetes phenotypes
As revealed by KEGG pathway analyses, both MODY and T1D were enriched pathways associated with highly perturbed genes of T2D, while MODY was also an enriched pathway associated with hub genes of T2D. Specifically, SLC2A2 (GLUT2), and IAPP genes were commonly enriched in MODY, while HLA-DRB4 and HLA-DRB5 genes were underlying the enrichment with T1D. A gene expression meta-analysis also revealed the existence of possible pleiotropic mechanisms manifest via common gene signatures (PGRMC1 and HADH) across different diabetes phenotypes (Mei et al. 2017).
Downregulation of SLC2A2 is associated with not only T2D (Solimena et al. 2018) but also neonatal diabetes (Sansbury et al. 2012) and early childhood diabetes (Alhaidan et al. 2020), suggesting a likely role in insulin secretion. Amylin (IAPP), a gluco-modulatory hormone co-expressed with insulin by pancreatic β cells, is downregulated in both T1D and advanced T2D (Abedini et al. 2013), while amylin agonists are considered novel therapeutic agents for treating diabetes (Sonne et al. 2021). Moreover, human amylin plays a protective role against autoimmune diabetes inducing CD4 + Foxp3 + regulatory T cells (Zhang et al. 2018). Downregulation of HLA-DRB4 in peripheral blood mononuclear cells is associated with T2D as well as dyslipidemia and periodontitis (Corbi et al. 2020), while a meta-analysis revealed that the lack of HLA-DRB5 increased T2D risk (Jacobi et al. 2020). Of note, both HLA-DRB4 and HLA-DRB5 are associated with β cell autoantibodies and T1D (Zhao et al. 2016), with previous studies reporting that both T1D and T2D share HLA class II locus components (Jacobi et al. 2020). Interestingly, two of the hub genes of T2D found in our study (MMP9, ARG1) have been found as hub genes of T1D in a previous analysis (Yang et al. 2020). Collectively, these findings support some degree of shared genetic architecture between T2D and other diabetes pathologies.
T2D as a disorder of insulin secretion and action
Downstream analyses provided insights into the characterization of T2D as a disorder of insulin secretion and action. We found that insulin secretion was the most significant KEGG pathway associated with hub genes, whereby two downregulated hub genes (SLC2A2, SNAP25) in T2D were underlying this enrichment. Zinc, the most significant HMDB metabolite associated with both highly perturbed genes and hub genes of T2D, is an essential element with key regulatory roles in insulin synthesis, storage, and secretion (Kim and Lee 2012). Other metabolomic markers associated with highly perturbed genes included magnesium which is necessary for insulin signaling (Piuri et al. 2021) as well as manganese which is involved in insulin synthesis and secretion (Chen et al. 2018). Together, these findings underscore the effects on insulin production and action as pivotal to T2D pathogenesis.
Pathophysiological manifestations of T2D
Apoptosis
Downstream analyses revealed that multiple GO and KEGG pathways associated with apoptosis, including intrinsic apoptotic signaling pathway, were enriched in T2D. It is known that hyperglycemia-induced β cell apoptosis, a hallmark in T2D progression, occurs via intrinsic pathways causing reduced islet mass and metabolic abnormalities (Wali et al. 2013). Hyperglycemia-induced apoptosis has been reported to occur in other sites such as renal cells (Jung et al. 2012) and coronary arteries (Kageyama et al. 2011), indicating a possible role in disease progression and the onset of complications.
Immunity
Downstream analyses also revealed that multiple immunity-related GO and KEGG pathways, encompassing both innate and humoral immune responses, were enriched in T2D. These included multiple ontologies involving neutrophils and antigen processing and presentation as well as severe immune reactions such as graft-versus-host disease and allograft rejection. Impaired immunity in T2D and consequent susceptibility to infections and complications is frequently observed (Berbudi et al. 2020). A deeper understanding of the genomics underlying impaired immunity in T2D might provide opportunities to personalize the management of comorbidities and pharmacotherapy.
COVID-19 and T2D
Epidemiological studies strongly suggest poorer prognosis of COVID-19 among people with T2D (Selvin and Juraschek, 2020), although underlying mechanisms are not well-understood (Apicella et al. 2020). Downstream analysis of highly perturbed genes and hub genes of T2D in the present study revealed a large number of enriched COVID-19-related gene sets, providing support for this putative link at a more granular level.
Over- and under-expression of genes in different tissues associated with T2D
Downstream analysis of GTEx profiles identified tissues that are likely to demonstrate under- and over-expression of DEGs associated with T2D. Findings indicate that adipose tissue tends to over-express marker genes of T2D, while these might be under-expressed in other tissues such as those of the nervous system. These findings have implications for biomarker discovery and can guide further research on tissues which should be explored for identifying DEGs.
Conclusions
Findings of this study contribute towards the understanding of the genetic basis of T2D, and further research is warranted to substantiate the molecular mechanisms underlying these findings which is fundamental to establishing precision T2D medicine initiatives. The proposed bioinformatics pipeline may have broader use as a judicious strategy to identify gene perturbations and pathophysiological mechanisms of other clinical conditions beyond T2D which ought to be validated in future research. Finally, this study describes an exemplary approach to applying comprehensive evidence synthesis using existing open-source gene expression data. Other researchers are encouraged to apply this methodology to obtain high-level evidence from existing multiple datasets, thereby getting the most value from existing bioinformatics sources.
Data availability
Data used in this study are freely available at the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) portal: https://www.ncbi.nlm.nih.gov/geo/
Code availability
All analytic codes are available on reasonable request from the corresponding author.
References
Abedini A, Schmidt AM (2013) Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett 587(8):1119–1127. https://doi.org/10.1016/j.febslet.2013.01.017
Alhaidan Y, Christesen HT, Højlund K, Al Balwi MA, Brusgaard K (2020) A novel gene in early childhood diabetes: EDEM2 silencing decreases SLC2A2 and PXD1 expression, leading to impaired insulin secretion. Mol Genet Genomics 295(5):1253–1262. https://doi.org/10.1007/s00438-020-01695-5
Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S (2020) COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol 8(9):782–792. https://doi.org/10.1016/S2213-8587(20)30238-2
Arroyo MN, Green JA, Cnop M, Igoillo-Esteve M (2021) tRNA biology in the pathogenesis of diabetes: role of genetic and environmental factors. Int J Mol Sci 22(2):496. https://doi.org/10.3390/ijms22020496
Aylward A, Chiou J, Okino ML, Kadakia N, Gaulton KJ (2018) Shared genetic risk contributes to type 1 and type 2 diabetes etiology. Hum Mol Genet ddy314 https://doi.org/10.1093/hmg/ddy314
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al (2013) NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 2013;41(Database issue):D991–5. https://doi.org/10.1093/nar/gks1193
Basile KJ, Guy VC, Schwartz S, Grant SF (2014) Overlap of genetic susceptibility to type 1 diabetes, type 2 diabetes, and latent autoimmune diabetes in adults. Curr Diab Rep 14(11):550. https://doi.org/10.1007/s11892-014-0550-9
Batista TM, Haider N, Kahn CR (2021) Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 64(5):994–1006. https://doi.org/10.1007/s00125-021-05415-5
Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R (2020) Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev 16(5):442–449. https://doi.org/10.2174/1573399815666191024085838
Bonnefond A, Boissel M, Bolze A, Durand E, Toussaint B, Vaillant E et al (2020) Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat Metab 2(10):1126–1134. https://doi.org/10.1038/s42255-020-00294-3
Buchberger E, Bilen A, Ayaz S, Salamanca D, Matas de Las Heras C, Niksic A, et al (2021) Variation in pleiotropic hub gene expression is associated with interspecific differences in head shape and eye size in Drosophila. Mol Biol Evol 2021:msaa335. https://doi.org/10.1093/molbev/msaa335
Calimlioglu B, Karagoz K, Sevimoglu T, Kilic E, Gov E, Arga KY (2015) Tissue-specific molecular biomarker signatures of type 2 diabetes: an integrative analysis of transcriptomics and protein-protein interaction data. OMICS 19(9):563–573. https://doi.org/10.1089/omi.2015.0088
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV et al (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. https://doi.org/10.1186/1471-2105-14-128
Chen P, Bornhorst J, Diana Neely M, Avila DS (2018) Mechanisms and disease pathogenesis underlying metal-induced oxidative stress. Oxid Med Cell Longev 2018:7612172. https://doi.org/10.1155/2018/7612172
Corbi SCT, de Vasconcellos JF, Bastos AS, Bussaneli DG, da Silva BR, Santos RA et al (2020) Circulating lymphocytes and monocytes transcriptomic analysis of patients with type 2 diabetes mellitus, dyslipidemia and periodontitis. Sci Rep 10(1):8145. https://doi.org/10.1038/s41598-020-65042-9
Davis S, Meltzer PS (2007) GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23(14):1846–1847. https://doi.org/10.1093/bioinformatics/btm254
Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30(1):207–210. https://doi.org/10.1093/nar/30.1.207
Flannick J, Johansson S, Njølstad PR (2016) Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat Rev Endocrinol 12(7):394–406. https://doi.org/10.1038/nrendo.2016.50
GTEx Consortium (2013) The Genotype-Tissue Expression (GTEx) project. Nat Genet 45(6):580–5. https://doi.org/10.1038/ng.2653
Harrison PF, Pattison AD, Powell DR, Beilharz TH (2019) Topconfects: a package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biol 20(1):67. https://doi.org/10.1186/s13059-019-1674-7
Hill DP, Smith B, McAndrews-Hill MS, Blake JA (2008) Gene Ontology annotations: what they mean and where they come from. BMC Bioinformatics 9(Suppl 5):S2. https://doi.org/10.1186/1471-2105-9-S5-S2
Idicula-Thomas S, Gawde U, Bhaye S, Pokar K, Bader GD (2020) Meta-analysis of gene expression profiles of lean and obese PCOS to identify differentially regulated pathways and risk of comorbidities. Comput Struct Biotechnol J 18:1735–1745. https://doi.org/10.1016/j.csbj.2020.06.023
Jacobi T, Massier L, Klöting N, Horn K, Schuch A, Ahnert P et al (2020) HLA class II allele analyses implicate common genetic components in type 1 and non-insulin-treated type 2 diabetes. J Clin Endocrinol Metab 105(3):dgaa027. https://doi.org/10.1210/clinem/dgaa027
Jung DS, Lee SH, Kwak SJ, Li JJ, Kim DH, Nam BY et al (2012) Apoptosis occurs differentially according to glomerular size in diabetic kidney disease. Nephrol Dial Transplant 27(1):259–266. https://doi.org/10.1093/ndt/gfr301
Kageyama S, Yokoo H, Tomita K, Kageyama-Yahara N, Uchimido R, Matsuda N et al (2011) High glucose-induced apoptosis in human coronary artery endothelial cells involves up-regulation of death receptors. Cardiovasc Diabetol 10:73. https://doi.org/10.1186/1475-2840-10-73
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Kim J, Lee S (2012) Effect of zinc supplementation on insulin resistance and metabolic risk factors in obese Korean women. Nutr Res Pract 6(3):221–225. https://doi.org/10.4162/nrp.2012.6.3.221
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44(W1):W90–W97. https://doi.org/10.1093/nar/gkw377
Le NQ, Do DT, Nguyen NT, Hung TN, Trang NT (2020) Identification of gene expression signatures for psoriasis classification using machine learning techniques. Medicine in Omics 1:100001. https://doi.org/10.1016/j.meomic.2020.100001
Lin Y, Li J, Wu D, Wang F, Fang Z, Shen G (2020) Identification of hub genes in type 2 diabetes mellitus using bioinformatics analysis. Diabetes Metab Syndr Obes 13:1793–1801. https://doi.org/10.2147/DMSO.S245165
Liu H, Qu Y, Zhou H, Zheng Z, Zhao J, Zhang J (2021) Bioinformatic analysis of potential hub genes in gastric adenocarcinoma. Sci Prog 104(1):368504211004260. https://doi.org/10.1177/00368504211004260
Liu Y, Gu HY, Zhu J, Niu YM, Zhang C, Guo GL (2019) Identification of hub genes and key pathways associated with bipolar disorder based on weighted gene co-expression network analysis. Front Physiol 10:1081. https://doi.org/10.3389/fphys.2019.01081
Liu Y, Yi Y, Wu W, Wu K, Zhang W (2019) Bioinformatics prediction and analysis of hub genes and pathways of three types of gynecological cancer. Oncol Lett 18(1):617–628. https://doi.org/10.3892/ol.2019.10371
Mei H, Li L, Liu S, Jiang F, Griswold M, Mosley T (2017) Tissue non-specific genes and pathways associated with diabetes: an expression meta-analysis. Genes (basel) 8(1):44. https://doi.org/10.3390/genes8010044
Meugnier E, Faraj M, Rome S, Beauregard G, Michaut A, Pelloux V et al (2007) Acute hyperglycemia induces a global downregulation of gene expression in adipose tissue and skeletal muscle of healthy subjects. Diabetes 56(4):992–999. https://doi.org/10.2337/db06-1242
Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q (2008) GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol 9(Suppl 1):S4. https://doi.org/10.1186/gb-2008-9-s1-s4
Nguyen TM, Shafi A, Nguyen T, Draghici S (2019) Identifying significantly impacted pathways: a comprehensive review and assessment. Genome Biol 20(1):203. https://doi.org/10.1186/s13059-019-1790-4
Palsgaard J, Brøns C, Friedrichsen M, Dominguez H, Jensen M, Storgaard H et al (2009) Gene expression in skeletal muscle biopsies from people with type 2 diabetes and relatives: differential regulation of insulin signaling pathways. PLoS ONE 4(8):e6575. https://doi.org/10.1371/journal.pone.0006575
Peña-Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C et al (2008) A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol 9(Suppl 1):S2. https://doi.org/10.1186/gb-2008-9-s1-s2
Piuri G, Zocchi M, Della Porta M, Ficara V, Manoni M, Zuccotti GV et al (2021) Magnesium in obesity, metabolic syndrome, and type 2 diabetes. Nutrients 13(2):320. https://doi.org/10.3390/nu13020320
Prada C, Lima D, Nakaya H (2020) MetaVolcanoR: gene expression meta-analysis visualization tool. R package version 1.4.0. 2020
Prasad RB, Groop L (2019) Precision medicine in type 2 diabetes. J Intern Med 285(1):40–48. https://doi.org/10.1111/joim.12859
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N et al (2019) Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 157:107843. https://doi.org/10.1016/j.diabres.2019.107843
Sansbury FH, Flanagan SE, Houghton JA, Shuixian Shen FL, Al-Senani AM, Habeb AM et al (2012) SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 55(9):2381–2385. https://doi.org/10.1007/s00125-012-2595-0
Scardoni G, Petterlini M, Laudanna C (2009) Analyzing biological network parameters with CentiScaPe. Bioinformatics 25(21):2857–2859. https://doi.org/10.1093/bioinformatics/btp517
Selvin E, Juraschek SP (2020) Diabetes epidemiology in the COVID-19 pandemic. Diabetes Care 43(8):1690–1694. https://doi.org/10.2337/dc20-1295
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504. https://doi.org/10.1101/gr.1239303
Solimena M, Schulte AM, Marselli L, Ehehalt F, Richter D, Kleeberg M et al (2018) Systems biology of the IMIDIA biobank from organ donors and pancreatectomised patients defines a novel transcriptomic signature of islets from individuals with type 2 diabetes. Diabetologia 61(3):641–657. https://doi.org/10.1007/s00125-017-4500-3
Sonne N, Karsdal MA, Henriksen K (2021) Mono and dual agonists of the amylin, calcitonin, and CGRP receptors and their potential in metabolic diseases. Mol Metab 46:101109. https://doi.org/10.1016/j.molmet.2020.101109
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB et al (2022) IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 183:109119. https://doi.org/10.1016/j.diabres.2021.109119
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://doi.org/10.1093/nar/gky1131
Takematsu E, Spencer A, Auster J, Chen PC, Graham A, Martin P et al (2020) Genome wide analysis of gene expression changes in skin from patients with type 2 diabetes. PLoS ONE 15(2):e0225267. https://doi.org/10.1371/journal.pone.0225267
Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, Chiou J et al (2018) Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med 15(9):e1002654. https://doi.org/10.1371/journal.pmed.1002654
Wali JA, Masters SL, Thomas HE (2013) Linking metabolic abnormalities to apoptotic pathways in Beta cells in type 2 diabetes. Cells 2(2):266–283. https://doi.org/10.3390/cells2020266
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P et al (2010) The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38(Web Server issue):W214-20
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R et al (2018) HMDB 4 0: the human metabolome database for 2018. Nucleic Acids Res 46(1):608-D617
Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL et al (2021) Gene set knowledge discovery with Enrichr. Curr Protoc 1(3):e90. https://doi.org/10.1002/cpz1.90
Yang S, Cao C, Xie Z, Zhou Z (2020) Analysis of potential hub genes involved in the pathogenesis of Chinese type 1 diabetic patients. Ann Transl Med 8(6):295. https://doi.org/10.21037/atm.2020.02.171
Yip L, Fuhlbrigge R, Alkhataybeh R, Fathman CG (2020) Gene expression analysis of the pre-diabetic pancreas to identify pathogenic mechanisms and biomarkers of type 1 diabetes. Front Endocrinol (lausanne) 11:609271. https://doi.org/10.3389/fendo.2020.609271
Zhang XX, Qiao YC, Li W, Zou X, Chen YL, Shen J et al (2018) Human amylin induces CD4+Foxp3+ regulatory T cells in the protection from autoimmune diabetes. Immunol Res 66(1):179–186. https://doi.org/10.1007/s12026-017-8956-5
Zhao LP, Alshiekh S, Zhao M, Carlsson A, Larsson HE, Forsander G et al (2016) Next-generation sequencing reveals that HLA-DRB3, -DRB4, and -DRB5 may be associated with islet autoantibodies and risk for childhood type 1 diabetes. Diabetes 65(3):710–718. https://doi.org/10.2337/db15-1115
Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14(2):88–98. https://doi.org/10.1038/nrendo.2017.151
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions KDS is supported by a PhD scholarship funded by the Australian government under Research Training Program (RTP). The funder was not involved in the design of the study; the collection, analysis, and interpretation of data; and writing the report and did not impose any restrictions regarding the publication of the report.
Author information
Authors and Affiliations
Contributions
KDS performed data acquisition, pre-processing, and curation of data, conducted the analyses, and wrote this manuscript. KDS, RTD, AF, and JE were responsible for study conceptualization and design and contributed to validating analyses, results, and interpretation and drafting the manuscript. DJ and AM contributed to study conceptualization and design and interpretation of data and critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not required being a review and secondary analysis of publicly available, deidentified data.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Silva, K., Demmer, R.T., Jönsson, D. et al. Highly perturbed genes and hub genes associated with type 2 diabetes in different tissues of adult humans: a bioinformatics analytic workflow. Funct Integr Genomics 22, 1003–1029 (2022). https://doi.org/10.1007/s10142-022-00881-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-022-00881-5