
Abstract
Reproductive functions are closely related to nutritional status. Recent studies suggest that adiponectin may be a hormonal link between them. Adiponectin is an adipocytokine, abundantly expressed in adipose tissues. It plays a dominant role in lipid and carbohydrate metabolism by stimulating fatty acid oxidation, decreasing plasma triglycerides, and increasing cells’ sensitivity to insulin and has direct antiatherosclerotic effects. The hormone is also postulated to play a modulatory role in the regulation of the reproductive system. The aim of this study was to identify differentially expressed genes (DE-genes) in response to adiponectin treatment of porcine luteal ovarian cells. The global expression of genes in the porcine ovary was investigated using the Porcine (V2) Two-color gene expression microarray, 4 × 44 (Agilent, USA). Analysis of the microarray data showed that 701 genes were differentially expressed and 389 genes showed a fold change greater than 1.2 (p < 0.05). Among this number, 186 genes were up-regulated and 203 were down-regulated. The list of DE-genes was used for gene ontology analyses. The biological process list was generated from up-regulated and down-regulated DE-genes. We found that up-regulated products of DE-genes take part in 30 biological processes and down-regulated products in 9. Analysis of the interaction network among DE-genes showed that adiponectin interacts with genes involved in important processes in luteal cells. These results provide a basis for future work describing the detailed interactions and relationships explaining local regulation of adiponectin actions in the ovary of pigs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the last two decades, the view on adipose tissue has changed from it being a passive reservoir for energy storage to a complex and highly active metabolic and endocrine organ. Adipose tissue is now known to express and secrete a variety of bioactive peptides, i.e., leptin, tumor necrosis factor alpha, interleukin 6, resistin, adipsin, and adiponectin (Kershaw and Flier 2004). Adiponectin is an adipocytokine, abundantly expressed in adipose tissues. It is also one of the most presented proteins in mammalian blood serum (2–25 μg/ml) (Kadowaki and Yamauchi 2005). The hormone is composed of 247 amino acids and has a molecular weight of about 28 kDa (Scherer et al. 1995). Adiponectin structurally belongs to the complement-1q family (Wong et al. 2004). As a result of post-translation modification, adiponectin circulates in blood in four different forms. The full-length peptide generates trimers, hexamers, and high-molecular-weight multimers and the globular fraction product of the proteolytic cleavage of the full-length peptide (Chandran et al. 2003).
Adiponectin’s physiological effects are dependent on receptor type 1 (AdipoR1) and type 2 (AdipoR2). AdipoR1 and AdipoR2 proteins in pigs have a molecular mass of 42 and 46 kDa, respectively (Chabrolle et al. 2007a). In humans and rodents, AdipoR1 is abundantly expressed in the skeletal muscle, whereas AdipoR2 is predominantly expressed in the liver (Yamauchi et al. 2003). Due to the extensive distribution of adiponectin receptors in the peripheral tissues and organs, adiponectin exerts pleiotropic effects on the regulation of energy homeostasis in organisms. It plays a dominant role in lipid and carbohydrate metabolism by stimulating fatty acid oxidation, decreasing plasma triglycerides, and increasing cells’ sensitivity to insulin and has direct antiatherosclerotic effects (Kadowaki and Yamauchi 2005). Adiponectin is also involved in the regulation of energy balance and body weight and additionally plays a modulatory role in the regulation of the reproductive system (Yamauchi et al. 2001; Kiezun et al. 2014; Maleszka et al. 2014a).
Reproductive functions are closely related to nutritional status. Recent studies suggest that adiponectin may be a hormonal link between them. The presence of adiponectin and its receptors was found at all levels of the hypothalamic-pituitary-gonadal (HPG) axis in the pig, human, rat, cow, and chicken (Ledoux et al. 2006; Chabrolle et al. 2007a, 2007b, 2009; Mailard et al. 2010).
In the hypothalamus, the presence of adiponectin receptors has been demonstrated in the neurons responsible for the secretion of gonadotropin-releasing hormone (GnRH) (Kaminski et al. 2014). Adiponectin reduces the secretion of GnRH in the GT1-7 cell line derived from murine GnRH-releasing neurons (Wen et al. 2008). The expression of adiponectin receptors was also found in the pituitary gland, which allows for the direct effect of the hormone on the secretion of the luteinizing hormone and follicle-stimulating hormone (Rodriguez-Pacheco et al. 2007; Kiezun et al. 2014). Another proof for the involvement of adiponectin in the regulation of the HPG axis is the demonstrated presence of adiponectin receptor protein in human and pig ovaries (Chabrolle et al. 2009; Maleszka et al. 2014a).
The systematically growing number of publications confirms the potential impact of adiponectin on the gene expression and secretion of hormones connected to reproductive functions. Ledoux et al. (2006) observed higher levels of prostaglandin-endoperoxide synthase 2 (PTGS2), prostaglandin E synthase (PTGES), and vascular endothelial growth factor (VEGF) gene expression in granulosa cells primed with adiponectin, an increase in the expression of the steroidogenic acute regulatory protein (StAR) gene, and a decrease in the expression of the P450 aromatase gene in granulosa cells derived from prepubertal gilts. Adiponectin was also found to affect the basal and stimulated secretion of steroid hormones, i.e., progesterone, estradiol, androstenedione, and testosterone, by ovarian cells of mature gilts (Maleszka et al. 2014a). These results undoubtedly confirm the involvement of adiponectin in the functioning of the reproductive system.
Despite the above, there is a lack of data concerning adiponectin’s influence on the transcriptome profile of the HPG axis cells, including ovarian cells. Knowledge about the impact of adiponectin on the functioning of the ovaries is fragmentary and needs more research based on microarray methods. The results of these studies may become a starting point for more detailed research clarifying these relationships. Thus, using the pig as a model, and a transcriptomic approach, the aim of this study was to identify differentially expressed genes during adiponectin treatment of porcine luteal cells.
Materials and methods
Experimental animals
All experiments were carried out in accordance with the principles of the ethical standards of the Animal Ethics Committee at the University of Warmia and Mazury in Olsztyn. The experimental material comprised mature gilts (Large White × Polish Landrace) from a private breeding farm, aged 7–8 months and weighing 130–140 kg. All of the gilts used in the experiment were on days 10 to 12 of the estrous cycle (n = 4). Females were monitored daily for estrous behavior in the presence of an intact boar. The day of the second estrous was designated as day 0 of the estrous cycle. The phase of the estrous cycle was also determined based on the ovarian morphology (Akins and Morrissette 1968). The ovaries were removed and placed on ice within minutes after slaughter. Subsequently, the ovaries from days 10 to 12 of the cycle were placed in cold PBS buffer and transported to the laboratory where luteal cells were isolated.
Isolation of the luteal cells
Luteal cells were isolated using the method described by Kaminski et al. (1999). Dissected corpora lutea were minced into small fragments and dispersed in F-12 medium (Sigma, USA) containing bovine serum albumin fraction V (BSA; 1 %; MP Biomedicals, USA) and antibiotics (Sigma, USA). Corpora lutea were enzymatically dissociated in 0.125 % trypsin solution (Biomed Lublin, Poland) (four to six times) at 37 °C, centrifuged (300×g, 10 min, 4 °C), and washed three times. Isolated luteal cells were filtered through the nylon mesh (40 μm in diameter) and resuspended in the fresh F-12 medium. The cells were counted using a hemocytometer, and their viability (∼90 %) was determined by 0.4 % trypan blue dye exclusion.
In vitro cultures of the luteal cells
Isolated luteal cells (1,500,000/1 mL medium) were resuspended in F-12 medium enriched with fetal calf serum (FCS; 20 %; Sigma USA), BSA (1 %) and antibiotics and preincubated in the 6-well plates for 48 h in a humidified incubator with 95 % air and 5 % CO2 atmosphere. The serum-containing medium was discarded, and the cells were washed using serum-free F-12 medium. After washing, the cells were cultured for 24 h in F-12 medium with BSA (1 %) with antibiotics, with or without (control) recombinant human adiponectin (BioVendor, USA) at the dose of 10 μg/ml. Cultures for each individual were performed in duplicate. The dose of adiponectin was equivalent to physiological concentration (Maleszka et al. 2014b). The alamarBlue test (MP Biomedicals, USA) revealed that the treatment did not affect the viability of the cultured cells. The alamarBlue test was performed as described by Rak and Gregoraszczuk (2008), with the modification of cell preincubation and proper incubation time (24 h each). The measurement of the absorbance was performed using an Infinite 200 PRO plate reader (Tecan Group, Switzerland) at wavelengths 570 and 600 nm.
Total RNA isolation and quality control
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, USA) and purified by on-column digestion of DNA with DNase I as recommended by the manufacturer (Qiagen, USA) to eliminate residual genomic DNA. The initial RNA quality and quantity were determined with spectrophotometry (Infinite 200 PRO plate reader with NanoQuant plates, Tecan Group, Switzerland). Subsequently, RNA quality was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies, USA) using RNA 6000 Nano Assay Kit (Agilent Technologies, USA). RNAs with an RNA integrity number (RIN) score between 8 and 10 were used for microarray and quantitative real-time PCR (qPCR) experiments. All RNAs were stored at −80 °C.
Microarray analysis
Total RNA was amplified and labeled using a Low Input Quick Amp, Two-Color kit (Agilent Technologies, USA) according to the manufacturer’s instructions. The sample and control RNAs were labeled with Cyanine-5 (Cy-5) and Cyanine-3 (Cy-3), respectively. Quantity and purity of cRNA were measured with Infinite 200 PRO plate reader with NanoQuant plates (Tecan Group, Switzerland). For each microarray, 825 ng of Cy-3 and 825 ng of Cy-5-labeled cRNA were mixed together. Dual-labeled cRNA was fragmented, mixed with hybridization buffer, and added to Porcine (V2) Gene Expression Microarray 4x44 (Agilent Technologies, USA) according to the manufacturer’s instructions. Two differentially labeled cRNA samples (obtained from treated and control samples) were placed on each array (n = 4, one slide) in a balanced block design with dye swaps (Supplementary Data 1). Hybridization was carried out at 60 °C for 17 h in an Agilent hybridization oven. Hybridized microarrays were dissociated form the hybridization chamber and washed. After washing, slides were scanned at 5 μm resolution with an Agilent’s High-Resolution C Microarray Scanner. The TIFF image generated was loaded into Feature Extraction Software (Agilent Technologies, USA) for feature data extraction and detailed analysis, background subtraction from features, and dye normalizations (linear and LOWESS).
Bioinformatic analysis
The expression data was loaded into GeneSpring GX 12 software (Agilent Technologies, USA) to determine which genes were differentially expressed between treated and control samples. Differentially expressed transcripts were identified by cutoff of fold change (FC) greater than 1.2 in up- or down-regulation and p value <0.05. The 1.2 FC cutoff was chosen based on other publications, which used the microarray technology (Franczak et al. 2014; Jeong et al. 2006; Kimsa et al. 2013). Student’s t test was used to compare the normalized fluorescence intense for adiponectin treated versus control samples. FC was calculated based on the mean values of the treated/control gene expression levels of four biological replicates. For genes with replicate probes on the array, which showed differential expression, the FC mean value of all the probes was calculated.
Gene ontology analysis
The list of differentially expressed genes (DE-genes) was uploaded into Database for Annotation, Visualization, and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov) to explore functional class scoring in the resulting gene list by means of GO term enrichment analysis (Huang et al. 2009). Significance levels were calculated following a modification of Fisher’s exact test.
Biological pathways analysis
DAVID was additionally used to explore the biological pathways enriched in the resulting gene list, using information from each individual gene and computing a total overrepresentation value for each pathway represented in the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg) and BioCarta (http://www.biocarta.com). Significance levels were calculated following a modification of Fisher’s exact test. As a complementary approach, Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/) bioinformatic tools were employed to explore the distribution of differentially expressed genes in well-reported canonical signal transduction or metabolic pathways integrating fold-change data from the microarray experiment and gene-to-gene or protein-to-protein interaction information generated from the literature databases. In this approach, statistical significance of pathway overrepresentation was established with respect to a null distribution constructed by permutations.
Interaction network of differentially expressed genes
IPA was used to examine molecular pathways. This software combines functional annotations of our differentially expressed genes and the corresponding bibliographic data to generate significant signaling pathways and regulation networks.
Real-time PCR validations
In order to confirm microarray results, nine genes were tested for validation by qPCR by using the same samples used for microarray studies. One microgram of total RNA samples (n = 4 for treated and n = 4 for the control) was reverse-transcribed into cDNA in a total volume of 20 \( \mu \)L with 0.5 \( \mu \)g oligo(dt)15 Primer (Roche, Germany) using the Omniscript RT Kit (Qiagen, USA) at 37 °C for 1 h and was terminated by incubation at 93 °C for 5 min. qPCR analysis was performed using PCR System 7300 and SYBR® Green PCR Master Mix (Applied Biosystems, USA). The PCR reaction included the following: 20 ng cDNA (all genes with the exception of nuclear receptor coactivator 3 (NCOA3 40 ng)), one of nine primer pairs (forward and reverse): angiotensin-converting enzyme (ACE)—500 nM, aldo-keto reductase family 1, member C1 (AKR1C1)—500 nM, prostaglandin-endoperoxide synthase 2/cyclooxygenase-2 (PTGS2/COX2)—500 nM, glutamine-fructose-6-phosphate aminotransferase 1 (GFPT1)—500 nM, interleukin 15 (IL15)—500 nM, NCOA3—250 nM, methylmalonyl-CoA mutase (MUT)—100 nM, nicotinamide phosphoribosyltransferase/visfatin (NAMPT)—300 nM, vascular endothelial growth factor α (VEGFα)—500 nM, 12.5 μl SYBR Green PCR Master Mix (Applied Biosystems, USA), and RNase free water to a final volume of 25 μl. Selected forward and reverse primers, as well as qPCR conditions, are presented in Table 1. Constitutively expressed genes, β-actin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were used as an internal control to verify the quantitative real-time PCR. To ensure that β-actin and GAPDH were suitable reference genes for this study, we revealed that there were no statistically significant differences in Ct values between the treated and control sample.
Results
Microarray data analysis
Description of differentially expressed genes
A table containing a complete list of genes that were differentially expressed between controls and the cells treated with 10 μg/ml of adiponectin is presented in Supplementary Data 2. The table includes a probe set name, gene ID, fold change, the direction of change (up or down), p value, as well as function and accession number. Out of these 701 genes, 389 genes were a fold change greater than 1.2 (p < 0.05); among this number, 186 genes were up-regulated and 203 were down-regulated.
Gene ontology analysis
GO term annotations were used to perform a functional categorization of the resulting list of differentially expressed gene analysis. Three GO categories (biological process, metabolic function, and cell component) were explored with DAVID bioinformatic tools for overrepresentation of specific GO terms. Significant results for the whole DE-gene list (up- and down-regulated; modified Fisher exact p < 0.1) are summarized in Supplementary Data 3. The most relevant results were obtained under the biological process category. Additionally, the biological process lists from up-regulated and down-regulated differentially expressed genes (DE-genes) were generated (Table 2). In the analysis conducted on the basis of the up-regulated DE-gene list, we observed a group of nine genes associated with GO terms related to macromolecular complex subunit organization (GO:0043933). Most of these are involved in the protein complex assembly (GO:000646), which suggests that adiponectin induces the formation of new protein complexes in luteal cells. Moreover, the same group of gene products is associated with the protein complex biosynthesis (GO:0070271), which indicates the involvement of adiponectin not only in the formation of protein complexes but also in the biosynthesis of new proteins. The next group of seven-gene products is involved in cytoskeleton organization (GO:0007010), including four engaged in the actin cytoskeleton organization (GO:0030036) process. The same group of four-gene products is involved in the actin filament-based process (GO:0030029). It seems that adiponectin may influence luteal cells via reorganization of their cytoskeleton. Another biological process modulated by the studied hormone is cell proliferation (GO:0008283). We have identified six up-regulated DE-genes related to this GO term. Another five-gene products are associated with blood circulation (GO:0008015), including three involved in the regulation of blood pressure (GO:0008217). Additionally, the next group of three-gene products is involved in the tissue remodeling, with two of them engaged in the blood vessel remodeling (GO:0001974). This suggests that the transport of nutrients and the removal of waste products from the highly metabolic luteal cells, regulated by the products of these two genes groups, may be affected by adiponectin. The adipokine increased the expression of five genes, whose products are involved in the cell response to hypoxia (GO:0001666). Another group of four genes is associated with GO term related to lipid localization (GO:0010876). It suggests that adiponectin may affect essential condition discharge of luteal cell steroidogenesis, which is the access to lipid. The next group of four-gene products is involved in the epithelium development (GO:0060429); three of these gene products are connected with epithelial cell differentiation (GO:0030855). Adiponectin has also an effect on the reproductive developmental processes (GO:0003006) by increasing the expression of four DE-genes. Three of these DE-genes affect gonad development (GO:0008406), development of primary sexual characteristic (GO:0045137), and reproductive structure development (GO:0048608). Another biological process worth emphasizing is the female pregnancy (GO:0007565), which is represented by three DE-genes. Moreover, three DE-genes are involved in the regulation of smooth muscle cell proliferation (GO:0048660), kidney development (GO:0001822), and nucleobase, nucleotide, and nucleic acid transport (GO:0015931). The last two significant results were related to the positive regulation of endothelial cell proliferation (GO:0001938) and glycosphingolipid metabolic process (GO:0006687). In the case of GO analysis of down-regulated DE-genes, the most significant group consisted of five genes, the products of which are involved in cell death (GO:0008219). Additionally, adiponectin decreased the expression of four genes connected to the GO term cell cycle process (GO:0022402). The adipokine down-regulated four DE-genes associated with the generation of precursor metabolites and energy (GO:0006091). Three of these genes are related to the energy reserve metabolic process (GO:0006112). Other processes affected by DE-genes are glutamine metabolic process (GO:0006541), cellular amide metabolic process (GO:0043603), and immunoglobulin-mediated immune response (GO:0016064), two DE-genes for each process. In summary, many up-regulated by adiponectin biological processes were related to the development and proliferation, whereas those connected to the cell death and catabolic processes were down-regulated. The above is consistent with the role of adiponectin as a factor promoting reproduction under proper nutritional conditions.
Biological pathway analysis
To generate biological pathway analysis, we used three different databases, which were KEGG, BioCarta, and IPA. The results are compared in Table 3. DAVID software generated three pathways based on KEGG database: purine metabolism (four DE-genes), renal cell carcinoma pathway (three DE-genes), and pantothenate and CoA biosynthesis (two DE-genes). In turn, BioCarta database generated one pathway, which was the transcription regulation by methyltransferase of CARM1. The second tool IPA software was used to survey the pathways overrepresented in our DE-gene list. The outcome of this analysis was a total of 16 pathways. The most significant was IL-8 signaling pathways (four DE-genes). Other pathways indicated by IPA tool were as follows: TR/RXR activation (three DE-genes), PPAR (three DE-genes), interleukins IL-6 (three DE-genes), and IL-10 signaling pathways (two DE-genes). Less significant pathways included i.a. nitric oxide signaling in the cardiovascular system (three DE-genes), LXR/RXR activation (three DE-genes), corticotropin-releasing hormone (three DE-genes), and pancreatic adenocarcinoma (three DE-genes) signaling, as well as ovarian cancer (three DE-genes). The differences between data obtained from IPA and DAVID are the result of different background to map the queried gene list, different search parameters, and algorithms used to define pathways in each database. In contrast to DAVID, IPA allows to find the pathways in the determined type of cells.
Network between differentially expressed genes
Differentially expressed genes containing the gene identifiers along with the corresponding fold changes were uploaded into the IPA software to annotate bio-functions as well as to construct and visualize molecular interaction networks. The IPA contains information from scientific publications regarding direct and indirect relationships between genes and proteins. Each identifier was mapped to its corresponding gene object in the IPA base. These genes, called focus genes, were overlaid onto a global molecular network in the IPA. Networks of these focus genes were then algorithmically generated based on their connectivity. A total of eight significant molecular networks were identified by Fisher’s exact test in the IPA software (Fig. 1a). The score was transformed from logP. Figure 1b, c showed two most significant networks, in which DE-genes were highlighted in red (up-regulated) and green (down-regulated). The first network (Fig. 1b) was built of 34 genes including 17 genes from the DE-gene list. The analysis indicated that DE-genes are involved in several bio-functions, which are cellular assembly and organization, cellular development, and cellular growth and proliferation. The second network (Fig. 1c) was constructed of 29 genes including 15 DE-genes that are involved in cell-mediated immune response, cellular development, and cellular function and maintenance. The third network was built of 28 genes and 15 DE-genes, which were also engaged into cellular and tissue development and cancer. The fourth network included 14 DE-genes within 26 genes, which are involved in cancer and gastrointestinal and infectious disease. The rest of the networks were built respectively of 22 genes (12 DE-Genes), 19 genes (11 DE-Genes), 14 genes (9 DE-Genes), and 7 genes (5 DE-Genes). Top bio-functions were as follows: cancer, cellular function and maintenance, molecular transport, cardiovascular diseases, connective tissue disorders, amino-acid metabolism, cellular development, cell death and survival, hematological diseases.

Interaction networks of selected genes. a Top network associations identified in microarray study; b the most significant molecular network by IPA pathway enrichment analysis; c the second most significant molecular network. Differentially expressed genes were highlighted in red (up-regulated) and green (down-regulated)
Real-time PCR validations
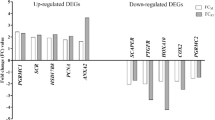
To validate DNA microarray data, nine genes with fold change greater than 1.2 were chosen for qPCR. The products of selected genes are involved in processes important for ovarian tissue functions, such as renin-angiotensin system (ACE), metabolism of lipids, carbohydrates, and protein (MUT, GFPT1, NAMPT), steroid hormone metabolism and action (AKR1C1, NCOA3), prostaglandin synthesis and metabolism (PTGS2), and interleukin and growth factor action (IL15, VEGFA). All changes in gene expression determined by DNA microarray were confirmed by qPCR (Fig. 2). All samples were amplified in duplicate. The specificity of amplification was tested at the end of the PCR by melting curve analysis. Levels of gene expression were calculated using the ΔΔCt method and normalized using the geometrical means of reference gene expression levels: β-actin and GAPDH. The Ct values for all non-template controls were under the detection threshold. Data are presented as means ± S.E.M. from four different observations. Differences between treated sample and control were analyzed by one-way ANOVA followed by least significant differences (LSD) post hoc test. Statistical analyses were performed using STATISTICA Software (StatSoft Inc., Tulsa, USA). Values of p < 0.05 were considered as statistically significant.

Results of microarray experiment validation with qPCR. Light grey bars indicate microarray fold change; dark grey bars indicate qPCR fold change. Data are presented as means ± S.E.M. from four different observations *p < 0.05; **p < 0.01
Discussion
The aim of this study was to determine for the first time the influence of adiponectin on the transcriptomic profile of luteal ovarian cells. An Agilent DNA microarray was used to identify the global gene expression profiles in the samples. In order to recognize the differentially expressed genes between the treated and non-treated luteal cells from the mid-luteal phase (days 10 to 12) of the estrous cycle, we extracted only those with a fold change above 1.2. The same type of microarray technology and cutoff 1.2 fold change was used by Franczak et al. (2014), Jeong et al. (2006), Kimsa et al. (2013). These allowed us to create a list of genes, which was used to describe gene ontology, biological pathways, and possible interaction networks.
We observed that 701 genes were differentially expressed. Among this number, the expression of 389 genes was altered by 1.2-fold in treated versus non-treated samples. Of them, 186 were up-regulated, whereas 203 were down-regulated. Other microarray analyses of the ovarian tissues conducted by Caetano et al. (2004) and Agca et al. (2006) indicated both quantitative and qualitative variations in the global gene expression. Agca et al. (2006) studied differences in the global gene expression between ovarian follicles before and after luteinization (43 up-regulated and 107 down-regulated genes). Caetano et al. (2004) observed differences in the various ovarian tissues gene expression during days 2–6 of the luteal phase between gilts line with the high-average oocyte production and randomly selected control line. The researchers identified 88 and 74 unique probes, representing 71 and 59 unique genes, which were differentially expressed between lines in the ovary and ovarian follicles of different size classes, respectively. These studies were not directly comparable with our data due to the differences in the experiment design, i.e., the used treatment, type of tested tissues, and the phase of the estrous cycle. However, they did observe changes in gene expression of similar magnitude to this study.
Previous studies demonstrated the impact of adiponectin on gene expression in the porcine ovary. However, the studies were limited to a predetermined group of genes and not to the entire transcriptome. Ledoux et al. (2006) showed the response of granulosa cells derived from prepubertal gilts to adiponectin. The authors observed higher levels of PTGS2 and VEGF gene expression in the cells primed with adiponectin. Ledoux et al. (2006) also indicated a higher level of PTGES and StAR gene expression and a decrease in the expression of the P450 aromatase gene, which is in agreement with our observations.
The analysis of gene ontology indicated that the expression of many genes changed under the influence of adiponectin. We noted a group of genes associated with several reproductive process GO terms, including reproductive developmental process (GO:0003006), female pregnancy (GO:0007565), gonad development (GO:0008406), reproductive structure development (GO:0048608), and development of primary sexual characteristics (GO:0045137). Our study indicated for the first time the role of adiponectin in the above-mentioned biological processes and identified genes potentially involved in them.
The subject of our research was the luteal cells isolated in the mid-luteal phase of the estrous cycle. The luteal phase begins with the formation of the corpus luteum (CL) and ends in either pregnancy or luteolysis. The mid-luteal phase is characterized by the highest activity of CL, an endocrine gland secreting estrogens and, most of all, significant amounts of progesterone. Adiponectin had an inhibitory effect on in vitro secretion of progesterone by porcine luteal cells on days 10 to 12 of the estrous cycle (Maleszka et al. 2014a). The effect of adiponectin on ovarian basal progesterone secretion was also suggested in studies on cows, rats, and humans (Chabrolle et al. 2009; Mailard et al. 2010; Lagaly et al. 2008). Moreover, the insulin-induced production of progesterone and estradiol by bovine granulosa cells was significantly decreased as a result of adiponectin treatment (Mailard et al. 2010). In the study with bovine theca interna cells, Lagaly et al. (2008) observed that adiponectin decreased LH and insulin-induced production of progesterone. In this study, we observed changes in the expression of genes which may be involved in the regulation of steroid hormone secretion under adiponectin’s influence. We noted an increase in the expression of the AKR1C1 gene. A product of this gene is an enzyme involved in the conversion of progesterone into its biologically inactive metabolite 20α-hydroxyprogesterone (Penning 1997). This enzyme has been detected, e.g., in the porcine ovary and uterus, rat ovary and placenta, and human uterus (Seo et al. 2011; Akinola et al. 1997; Shiota et al. 1993; Zhang et al. 2000). Moreover, according to our results, AKR1C1 is a part of two-gene groups associated with macromolecular complex subunit organization (GO:0043933) and lipid localization (GO:0010876). However, studies of other researchers indicated these functions only in hepatocytes (Stolz et al. 1993). Another gene down-regulated by adiponectin which may be involved in progesterone regulation is IL1β. Studies on the effects of IL1β on progesterone production have been inconsistent. IL1β has been reported to suppress progesterone synthesis in the CL of rats (Hurwitz et al. 1997), to stimulate progesterone synthesis in human and pig luteal cells (Miceli et al. 2003; Zmijewska et al. 2013), or to have no effect in bovine luteal cells (Nishimura et al. 2004). The above suggests that there are species-dependent differences in the effects of IL1β on steroidogenesis. Adiponectin may also negatively regulate progesterone secretion via an observed decrease in the expression of visfatin. Earlier studies demonstrated that recombinant human visfatin (10 ng/ml) significantly increased IGF-1-induced progesterone and estradiol secretion in both primary human granulosa cells and the human ovarian granulosa-like tumor cell line (Reverchon et al. 2013).
Additionally, we found that NCOA3 and cAMP-response element binding protein (CREBBP) genes, which are involved in the regulation of steroid action, were up-regulated by adiponectin. NCOA3 interacts with numerous nuclear hormone receptors, like estrogen receptor alpha, thyroid hormone receptor, and progesterone receptor, to enhance their transcriptional activation (Bouras et al. 2001). Genetic disruption in mice results in a pleiotropic phenotype showing dwarfism, delayed puberty, reduced female reproductive function, and blunted mammary gland development (Xu et al. 2000). NCOA3 associates with the transcription factor CREBBP and possesses histone acetyltransferase activity (Chen et al. 1997). It plays a key role in creating a multisubunit coactivator complex, which probably acts via remodeling of chromatin. The CREBBP gene is ubiquitously expressed and is involved in the transcriptional coactivation of many different transcription factors. This gene is known to play critical roles in embryonic development, growth control, and homeostasis by coupling chromatin remodeling to transcription factor recognition (Ogryzko et al. 1996). It seems that adiponectin may have an indirect effect on the processes involved in the coactivation of different nuclear receptors, through changes in the expression of these two genes.
Another group of genes affected by adiponectin comprises those connected to prostaglandin synthesis. Prostaglandins have an important role in the regulation of the estrous cycle and successful implantation in pigs (Spencer and Bazer 2004). PTGS and PTGS2 are responsible for the formation of prostanoids, including prostaglandins (Sawdy et al. 2000; Charpigny et al. 1997). In our study, we noted the up-regulation of PTGS2 caused by adiponectin treatment. This may have an implication in reproductive events such as ovulation, luteolysis, implantation, and parturition. Previous research noted an increase in both enzymes’ mRNAs content after stimulation with adiponectin (Ledoux et al. 2006). Adipokine may have an influence on female pregnancy (GO:0007565), by an increase in the expression of PTGS2, chloride intracellular channel 5 (CLIC5), and the placental growth factor (PGF) gene. The observed expression of PTGS2 in the ovary may suggest the autocrine action of prostaglandins, including prostaglandins E and F2α, on luteinization and luteolysis of ovarian cells. Another gene involved in this process, CLIC5, is involved in chloride ion transport within various subcellular compartments (Berryman and Bretscher 2000). CLIC5 appears to be an important component of many actin-rich structures in cells. Placenta growth factor (PGF) is another gene affected by adiponectin. This suggests that this adipokine may regulate cell proliferation (GO:0008283) as well as endothelial cell proliferation (GO:0001938), via PGF expression modulation. PGF is a key molecule in angiogenesis and vasculogenesis during embryogenesis. The main source of PGF during pregnancy is the placental trophoblast. It stimulates the proliferation, migration, and activation of endothelial cells (Vuorela et al. 1997; Maglione et al. 1991). Adiponectin is also reported to have angiogenic activity (Ouchi et al. 2004) and therefore could be essential in the establishment of the vascular system in the ovary and potentially in the developing placenta, presumably by the stimulation of angiogenic factors, such as PGF.
Knowledge of the effects of adiponectin on the development of the ovaries is limited mainly to oocytes’ maturation and vascularization (Chappaz et al. 2008; Ledoux et al. 2006). Adiponectin addition to the medium induced meiotic maturation of oocytes derived from large follicles, but not those derived from small follicles (Chappaz et al. 2008). The growth and regression of ovarian tissues are accompanied by equally rapid changes in their vascular vessel network. The main role in ovarian angiogenesis may be played by VEGF, which stimulates vessel growth and increases endothelial permeability, thus favoring the uptake of nutrients by the developing tissues (Gomez et al. 2002). We observed an up-regulatory effect of adiponectin on this gene expression, which may suggest that the adipokine stimulates the above process. The above suggest that adiponectin may affect blood vessel remodeling (GO:0001974) and positive regulation of endothelial cell proliferation (GO:0001938) via VEGF expression modulation. In the group of genes involved in ovary development, we also noted OSR1 and SF1. These genes were up-regulated by adiponectin in this study. OSR1 is a transcription factor which participates in the normal development of body parts. In mutant mouse embryos with an inactive OSR1 gene, the complete agenesis of adrenal glands, metanephric kidneys, and gonads as well as defects in pericardium formation were observed (Wang et al. 2005). The SF1 gene encodes a nuclear pre-mRNA splicing factor. The SF1 protein is a critical regulator of reproduction, modulating the transcription of key genes involved in sexual development and reproduction. Its targets include genes at every level of the HPG axis, as well as many genes involved in gonadal and adrenal steroidogenesis (Jamesson 2004). Mice with granulosa cell-specific conditional knockout of SF1 are sterile and have fewer follicles, a lack of corpora lutea, and hemorrhagic cysts (Cordts et al. 2011). These characteristics indicate the important role of SF1 in the development of ovaries, which directly determines the proper functioning of this structure. This may suggest that adiponectin is also responsible for the proper development of reproductive organs. Also, the same group, i.e., VEGF, OSR1, and SF1 genes, was identified to be involved in another two biological processes: reproductive structure development (GO:0048608) and the development of primary sexual characteristics (GO:0045137). Changes in the plasma adiponectin concentration during sexual maturation seem to be significant evidence for its involvement in the above processes (Böttner et al. 2004). Our study provides evidence that adiponectin may affect sexual development via an influence on the expression of, e.g., VEGF, OSR1, and SF1 genes.
In conclusion, our study was the first experiment to demonstrate the effect of adiponectin on global gene expression in the cells of the corpora lutea during the mid-luteal phase. We identified a group of genes connected to reproductive system function whose expression was affected by the studied hormone. These results provide a basis for future work describing the detailed interactions and relationships explaining local regulation of adiponectin actions in the ovary of pigs.
References
Agca C, Ries JE, Kolath SJ, Kim JH, Forrester LJ, Antoniou E, Whitworth KM, Mathialagan N, Springer GK, Prather RS, Lucy MC (2006) Luteinization of porcine preovulatory follicles leads to systematic changes in follicular gene expression. Reprod (Camb) 132:133–145
Akinola LA, Poutanen M, Vihko R, Vihko P (1997) Expression of 17b-hydroxysteroid dehydrogenase type 1 and type 2, p450 aromatase, and 20a-hydroxysteroid dehydrogenase enzymes in immature, mature, and pregnant rats. Endocrinology 138:2886–2892
Akins EL, Morrissette MC (1968) Gross ovarian changes during estrous cycle of swine. Am J Vet Res 29(10):1953–1957
Berryman M, Bretscher A (2000) Identification of a novel member of the chloride intracellular channel gene family (CLIC5) that associates with the actin cytoskeleton of placental microvilli. Mol Biol Cell 11(5):1509–1521
Blitek A, Waclawik A, Kaczmarek MM, Stadejek T, Pejsak Z, Ziecik AJ (2006) Expression of cyclooxygenase-1 and −2 in the porcine endometrium during the oestrous cycle and early pregnancy. Reprod Domest Anim 41:251–257
Böttner A, Kratzsch J, Müller G, Kapellen TM, Blüher S, Keller E, Blüher M, Kiess W (2004) Gender differences of adiponectin levels develop during the progression of puberty and are related to serum androgen levels. J Clin Endocrinol Metab 89(8):4053–4061
Bouras T, Southey MC, Venter DJ (2001) Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu1. Cancer Res 61:903–907
Caetano AR, Johnson RK, Ford JJ, Pomp D (2004) Microarray profiling for differential gene expression in ovaries and ovarian follicles of pigs selected for increased ovulation rate. Genetics 168(3):1529–1537
Chabrolle C, Tosca L, Dupont J (2007a) Regulation of adiponectin and its receptors in rat ovary by human chorionic gonadotropin treatment and potential involvement of adiponectin in granulosa cell steroidogenesis. Reproduction 133:719–731
Chabrolle C, Tosca L, Crochet S, Tesseraud S, Dupont L (2007b) Expression of adiponectin and its receptors (AdipoR1 and AdipoR2) in chicken ovary: potential role in ovarian steroidogenesis. Domest Anim Endocrinol 33:480–487
Chabrolle C, Tosca L, Rame C, Lecomte P, Royere D, Dupont J (2009) Adiponectin increases insulin-like growth factor I-induced progesterone and estradiol secretion in human granulose cells. Fertil Steril 92:1988–1996
Chandran M, Phillips SA, Ciaraldi T, Henry R (2003) Adiponectin: more than just another fat cell hormone? Diabetes Care 26:2442–2450
Chappaz E, Albornoz MS, Campos D, Che L, Palin MF, Murphy BD, Bordignon V (2008) Adiponectin enhances in vitro development of swine embryos. Domest Anim Endocrinol 35:198–207
Charpigny G, Reinaud P, Tamby JP, Creminon C, Guillomot M (1997) Cyclooxygenase-2 unlike cyclooxygenase-1 is highly expressed in ovine embryos during the implantation period. Biol Reprod 57:1032–1040
Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/P300. Cell 90:569–580
Cordts EB, Christofolini DM, Dos Santos AA, Bianco B, Barbosa CP (2011) Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet 283:635–643
Franczak A, Wojciechowicz B, Kolakowska J, Zglejc K, Kotwica G (2014) Transcriptomic analysis of the myometrium during peri-implantation period and luteolysis - the study on the pig model. Funct Integr Genomics 14(4):673–682
Gomez R, Simon C, Remohi J, Pellicer A (2002) Vascular endothelial growth factor receptor-2 activation induces vascular permeability in hyperstimulated rats, and its effect is prevented by receptor blockade. Endocrinology 143:4339–4348
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Hurwitz A, Finci-Yeheskel F, Yagel S, Shimonovitz S, Laufer N, Adashi EY, Mayer M (1997) Interleukin-1b inhibits progesterone accumulation in rat corpora luteal cell cultures in a mechanism dissociated from its effects on nitric oxide and prostaglandin e accumulation. Mol Cell Endocrinol 133:41–48
Jamesson JL (2004) Editorial: of mice and men: the tale of steroidogenic factor-1. The J Clin Endocrinol Metab 89(12):5927–5929
Jeong JW, Kwak I, Lee KY, White LD, Wang XP, Brunicardi FC, O’Malley BW, DeMayo FJ (2006) The genomic analysis of the impact of steroid receptor coactivators ablation on hepatic metabolism. Mol Endocrinol 20(5):1138–1152
Kadowaki T, Yamauchi T (2005) Adiponectin and adiponectin receptors. Endocr Rev 26:439–451
Kaminski T, Siawrys G, Okrasa S, Przala J (1999) Action of the opioid agonist FK 33–824 on porcine small and large luteal cells from the mid-luteal phase: effect on progesterone cAMP, cGMP and inositol phosphate release. Anim Reprod Sci 56(3–4):245–257
Kaminski T, Smolinska N, Maleszka A, Kiezun M, Dobrzyn K, Czerwinska J, Szeszko K, Nitkiewicz A (2014) Expression of adiponectin and its receptors in the porcine hypothalamus during the oestrous cycle. Reprod Domest Anim 49(3):378–386
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89(6):2548–2556
Kiezun M, Smolinska N, Maleszka A, Dobrzyn K, Szeszko K, Kaminski T (2014) Adiponectin expression in the porcine pituitary during the estrous cycle and its effect on LH and FSH secretion. Am J Physiol Endocrinol Metab 307(11):1038–1046
Kimsa MC, Strzalka-Mrozik B, Kimsa MW, Kruszniewska-Rajs C, Gola J, Adamska J, Rajs A, Mazurek U (2013) Porcine endogenous retrovirus infection changes the expression of inflammation-related genes in lipopolysaccharide-stimulated human dermal fibroblasts. Ann Transpl 25(18):576–586
Lagaly DV, Aad PY, Grado-Ahuir JA, Hulsey LB, Spicer LJ (2008) Role of adiponectin in regulating ovarian theca and granulosa cell function. Mol Cell Endocrinol 284(1–2):38–45
Ledoux S, Campos D, Lopes F, Dobias-Goff M, Palin M, Murphy B (2006) Adiponectin induces periovulatory changes in ovarian follicular cells. Endocrinology 147:5178–5186
Liu M, Fang L, Tan C, Long T, Chen H, Xiao S (2011) Understanding streptococcus suis serotype 2 infection in pigs through a transcriptional approach. BMC Genomics 12:253
Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG (1991) Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci U S A 88:9267–9271
Mailard V, Uzbekova S, Guignot F, Perreau C, Rame C, Coyral-Castel S, Dupont J (2010) Effect of adiponectin on bovine granulosa cell steroidogenesis, oocyte maturation and embryo development. Reprod Biol Endocrinol 10:8–23
Maleszka A, Smolinska N, Nitkiewicz A, Kiezun M, Chojnowska K, Dobrzyn K, Szwaczek H, Kaminski T (2014a) Adiponectin expression in the porcine ovary during the oestrous cycle and its effect on ovarian steroidogenesis. Int J Endocrinol 2014:957076
Maleszka A, Smolinska N, Nitkiewicz A, Kiezun M, Dobrzyn K, Czerwinska J, Szeszko K, Kaminski T (2014b) Expression of adiponectin receptors 1 and 2 in the ovary and concentration of plasma adiponectin during the oestrous cycle of the pig. Acta Vet Hung 62(3):386–396
Miceli F, Tropea A, Minici F, Navarra P, Lanzone A, Apa R (2003) Interleukin-1b stimulates progesterone production by in vitro human luteal cells: evidence of a mediatory role of prostaglandins. J Clin Endocrinol Metab 88(6):2690–2694
Naranjo V, Hofle U, Vicente J, Martin MP, Ruiz-Fons F, Gortazar C, Kocan KM, de la Fuente J (2006) Genes differentially expressed in oropharyngeal tonsils and mandibular lymph nodes of tuberculous and nontuberculous european wild boars naturally exposed to Mycobacterium Bovis. FEMS Immunol Med Microbiol 46:298–312
Nishimura R, Bowolaksono A, Acosta TJ, Murakami S, Piotrowska K, Skarzynski DJ, Okuda K (2004) Possible role of interleukin-1 in the regulation of bovine corpus luteum throughout the luteal phase. Biol Reprod 71(5):1688–1693
Nitkiewicz A, Smolinska N, Przala J, Kaminski T (2010) Expression of orexin receptors 1 (OX1R) and 2 (OX2R) in the porcine ovary during the oestrous cycle. Regul Pept 165(2–3):186–190
Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators P300 and CBP are histone acetyltransferases. Cell 87(5):953–959
Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K (2004) Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and akt signaling in endothelial cells. J Biol Chem 279:1304–1309
Palin MF, Labrecque B, Beaudry D, Mayhuea M, Bordignon V, Murphy BD (2008) Visfatin expression is not associated with adipose tissue abundance in the porcine model. Domest Anim Endocrinol 35:58–73
Penning TM (1997) Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr Rev 18(3):281–305
Rak A, Gregoraszczuk EL (2008) Modulatory effect of ghrelin in prepubertal porcine ovarian follicles. J Physiol Pharmacol 59(4):781–793
Ramsay TG, Caperna TJ (2009) Ontogeny of adipokine expression in neonatal pig adipose tissue. Comp Biochem Physiol B Biochem Mol Biol 152:72–78
Reverchon M, Cornuau M, Cloix L, Rame C, Guerif F, Royere D, Dupont J (2013) Visfatin is expressed in human granulosa cells: regulation by metformin through AMPK/SIRT1 pathways and its role in steroidogenesis. Mol Hum Reprod 19(5):313–326
Rodriguez-Pacheco F, Martinez-Fuentes A, Tovar S, Pinilla L, Tena-Sempere M, Dieguez C, Castaño J, Malagon M (2007) Regulation of pituitary cell function by adiponectin. Endocrinology 148(1):401–410
Sawdy RJ, Slater DM, Dennes WJ, Sullivan MHF, Bennetta PR (2000) The roles of the cyclo-oxygenases types one and two in prostaglandin synthesis in human fetal membranes at term placenta. Placenta 21:54–57
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749
Seo KS, Naidansuren P, Kim SH, Yun SJ, Park JJ, Sim BW, Park CW, Nanjidsuren T, Kang MH, Seo H, Ka H, Kim NH, Hwang SY, Yoon JT, Yamanouchi K, Min KS (2011) Expression of Aldo-keto reductase family 1 member C1 (AKR1C1) gene in porcine ovary and uterine endometrium during the estrous cycle and pregnancy. Reprod Biol Endocrinol 9:139
Shiota K, Seong HH, Noda K, Hattori N, Ikeda A, Ogura A, Itagaki S, Takahashi M, Ogawa T (1993) 20α-Hydroxysteroid dehydrogenase activity in rat placenta. Endocr J 40(6):673–681
Spagnuolo-Weaver M, Fuerst R, Campbell ST, Meehan BM, Mcneilly F, Adair B, Allan G (1999) A fluorimeter-Based RT-PCR method for the detection and quantitation of porcine cytokines. J Immunol Methods 230:19–27
Spencer TE, Bazer FW (2004) Conceptus signals for establishment and maintenance of pregnancy. Reprod Biol Endocrinol 2:49
Stolz A, Hammond L, Lou H, Takikawa H, Ronk M, Shively JE (1993) cDNA cloning and expression of the human hepatic bile acid-binding protein. J Biol Chem 268(14):10448–10457
Vuorela P, Hatva E, Lymboussaki A, Kaipainen A, Joukov V, Persico MG, Alitalo K, Halmesmäki E (1997) Expression of vascular endothelial growth factor and placenta growth factor in human placenta. Biol Reprod 56(2):489–494
Wang Q, Lan Y, Cho ES, Maltby KM, Jiang R (2005) Odd-Skipped Related 1 (Odd1) is an essential regulator of heart and urogenital development. Dev Biol 288(2):582–594
Wang X, Chen J, Liu H, Xu Y, Wang X, Xue C, Yu D, Jiang Z (2008) The pig p160 co-activator family: full length cDNA cloning, expression and effects on intramuscular fat content in longissimus dorsi muscle. Domest Anim Endocrinol 35:208–216
Wen JP, Lv WS, Yang J, Nie AF, Cheng XB, Yang Y, Ge Y, Li XY, Ning G (2008) Globular adiponectin inhibits GnRH secretion from GT1-7 hypothalamic GnRH neurons by induction of hyperpolarization of membrane potential. Biochem Biophys Res Commun 371:756–761
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF (2004) A family Of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci U S A 101(28):10302–10307
Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O’Malley BW (2000) The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A 97(12):6379–6384
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7(8):941–9466
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769
Zhang Y, Dufort I, Rheault P, Luu-The V (2000) Characterization of a human 20alpha-hydroxysteroid dehydrogenase. J Mol Endocrinol 25(2):221–228
Zmijewska A, Franczak A, Kotwica G (2013) The interleukin-1β system in the corpora lutea of pigs during early pregnancy and the estrous cycle. J Reprod Immunol 98:62–68
Acknowledgment
This research was supported by the National Science Centre (project no. 2011/01/B/NZ4/01596).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szeszko, K., Smolinska, N., Kiezun, M. et al. The influence of adiponectin on the transcriptomic profile of porcine luteal cells. Funct Integr Genomics 16, 101–114 (2016). https://doi.org/10.1007/s10142-015-0470-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-015-0470-z