
Abstract
Background
Gastric cancer (GC) is histologically a very heterogeneous disease, and the temporal development of different histological phenotypes remains unclear. Recent studies in lung and ovarian cancer suggest that KRAS activation (KRASact) can influence histological phenotype. KRASact likely results from KRAS mutation (KRASmut) or KRAS amplification (KRASamp). The aim of the study was to investigate whether KRASmut and/or KRASamp are related to the histological phenotype in GC.
Methods
Digitized haematoxylin/eosin-stained slides from 1282 GC resection specimens were classified according to Japanese Gastric Cancer Association (JGCA) and the Lauren classification by at least two observers. The relationship between KRAS status, predominant histological phenotype and clinicopathological variables was assessed.
Results
KRASmut and KRASamp were found in 68 (5%) and 47 (7%) GCs, respectively. Within the KRASmut and KRASamp cases, the most frequent GC histological phenotype was moderately differentiated tubular 2 (tub2) type (KRASmut: n = 27, 40%; KRASamp: n = 21, 46%) or intestinal type (KRASmut: n = 41, 61%; KRASamp: n = 23, 50%). Comparing individual histological subtypes, mucinous carcinoma displayed the highest frequency of KRASmut (JGCA: n = 6, 12%, p = 0.012; Lauren: n = 6, 12%, p = 0.013), and KRASamp was more frequently found in poorly differentiated solid type (n = 12, 10%, p = 0.267) or indeterminate type (n = 12, 10%, p = 0.480) GC. 724 GCs (57%) had intratumour morphological heterogeneity.
Conclusions
This is the largest GC study investigating KRAS status and histological phenotype. We identified a relationship between KRASmut and mucinous phenotype. The high level of intratumour morphological heterogeneity could reflect KRASmut heterogeneity, which may explain the failure of anti-EGFR therapy in GC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is histologically a very heterogeneous disease, and this is reflected in the numerous proposed histological classification schemes [1]. The temporal development of different histological phenotypes in GC remains unclear. Recent studies suggest that Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) activation and downstream signalling can impact on the properties and functions of the tumour microenvironment [2], and thus may influence histological phenotype. Likely mechanisms of KRAS activation include KRAS mutation (KRASmut) and KRAS amplification (KRASamp) [3].
Mutations in KRAS have been identified in many human cancers and result in the constitutive activation of KRAS and the receptor tyrosine kinase (RTK) pathway [4]. The frequency of KRASmut is variable across different cancer types, with the highest frequency in pancreatic cancer (90%) followed by colon (34.6%), lung (16.5%) and ovarian (11%) cancer and the lowest frequencies in cervical (6.6%), prostate (5%) and oesophageal cancer (2%) [5]. In a review of the literature we identified, on average, only 6.5% of GC have a KRASmut [6]. In colorectal cancer, routine testing for KRASmut is now implemented as a predictor of response to anti-epidermal growth factor receptor (EGFR) therapy [7].
Several studies have demonstrated a relationship between KRASmut status and histological phenotype in lung and ovarian cancer. In the subgroup of invasive mucinous adenocarcinoma of the lung, KRAS is mutated in up to 86% of cases [8]. In ovarian cancer, KRASmut has been identified in almost all cases with a mucinous histological phenotype [9]. The relationship between KRASmut status and histological phenotype in GC remains to be clarified [6].
The reported frequency of KRASamp is 1–9% in GC [10,11,12,13,14,15,16]. There are no reports of a relationship between KRAS DNA copy number and histological phenotype in other cancer types and in GC it has not been investigated in a large study. There is increasing recognition of the clinical importance of KRASamp in GC. KRASamp is also associated with a worse survival [3, 10, 12], whereas KRASmut do not appear to influence survival of GC patients [17].
Recently, image analysis on lung cancer haematoxylin and eosin (H&E) stained sections using deep learning was predictive of mutation status [18], thus suggesting that morphological phenotype is reflective of molecular phenotype. Investigating the relationship between KRAS activation by KRASmut and/or KRASamp and histological phenotype may provide some insight into gastric adenoma–carcinoma sequence progression and the origin of histological heterogeneity. Based on the studies in lung and ovarian cancer, we hypothesise that KRAS activation influences histological phenotype and is associated with a mucinous phenotype in GC. This would suggest that KRAS activation is an early event in GC, occurring before the phenotype is determined.
The aim of this multicentre GC study was to investigate the relationship of KRAS activation status (KRASmut and/or KRASamp) with the histological phenotype in a large series of GCs from UK, Japan and The Cancer Genome Atlas (TCGA). In addition, the relationship between KRAS status, clinicopathological variables, survival and microsatellite instability status was assessed.
Material and methods
Patients
Kanagawa Cancer Center Hospital (KCCH), Yokohama, Japan
This cohort included 250 patients with TNM stage II/III GC who underwent potentially curative surgery at Kanagawa Cancer Center Hospital (Yokohama, Japan) between 2001 and 2010. One hundred and six (43%) patients were treated with surgery alone, 108 (43%), 22 (9%), 14 (6%) patients received S-1, tegafur–uracil or S-1 combined with other cytotoxic drug therapy, respectively. Demographical, clinical and pathological data were retrieved from hospital records. The study was approved by the Local Research Ethics Committee.
Leeds Teaching Hospitals NHS Trust (LTHT), Leeds, UK
This cohort included 277 patients with GC who underwent potentially curative surgery at the Department of Surgery, Leeds General Infirmary (Leeds, UK), between 1970 and 2004. Seven (3%) patients were treated by chemotherapy followed by surgery and the remaining 270 (98%) by surgery alone. Clinical and pathological data were retrieved from histopathology reports, electronic patient hospital records and the Northern and Yorkshire Cancer Registry. The study was approved by the Local Research Ethics Committee (LREC no. CA01/122).
The Cancer Genome Atlas
The TCGA stomach adenocarcinoma (STAD) clinicopathological and molecular dataset of 295 patients was obtained from the publically available TCGA database portal [19].
Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology (TMGH), Tokyo, Japan
This cohort included 420 patients with 460 GC who were treated by surgery in the Tokyo Metropolitan Geriatric Hospital between 2000 and 2008. Three hundred and eighty patients had single carcinoma, and 36 had 2 or more carcinomas. Patients with Lynch syndrome were excluded from the current study. None of the patients underwent neoadjuvant chemotherapy. Histopathological examination and medical research were performed with informed written consent by the patients, and this work was approved by the ethics committee of the Tokyo Metropolitan Geriatric Hospital (#230,225, R16-23).
Histopathological classification
pT and pN stages were reported according to 7th edition of the UICC TNM classification for GC [20].
In all cohorts, H&E stained formalin fixed paraffin embedded (FFPE) tissue sections from the resection specimens were reviewed. In the KCCH and LTHT cohorts, H&E stained slides were scanned at 40 × magnification using an Aperio AT2 scanner for review. In the TCGA cohort, H&E stained slides were viewed online using the cancer digital slide archive (https://cancer.digitalslidearchive.net/). In the TMGH cohort, classification was performed using the glass slides.
Histological classification according to JGCA scheme was performed [21]. Mucinous carcinoma were defined as tumour cells located in mucinous pools comprising an area greater than 50% of the total tumour. GC were classified as signet-ring cell carcinoma when signet-ring cells were present in more than 50% of the tumour volume. In cases where more than one histological phenotype was identified, the most predominant phenotype was recorded, and these GCs were categorised as heterogeneous. JGCA classification was converted to Lauren classification [22] according to Table 1. As there is no Lauren classification for mucinous GC, we retained mucinous carcinomas as a separate category to distinguish them from other histological types.
DNA extraction
The area with the highest tumour cell density was identified on H&E stained sections and the whole tumour area, irrespective of subregions with different histological phenotypes was microdissected after staining with Shandon instant haematoxylin (Thermo Scientific, Cheshire, UK) using a sterile surgical blade. Tumour DNA from FFPE material was extracted from KCCH and LTHT GCs using the QIAmp DNA Micro Kit (Qiagen, Hilden, Germany) as previously described [23]. DNA concentration was measured by ND-100 Spectrophotometer (Labtech International) and samples were diluted using Tris-EDTA buffer. In the TMGH cohort, DNA was extracted using a phenol–chloroform procedure as described previously [24].
KRAS gene copy number and data analyses
KRAS copy number status was investigated in KCCH, LTHT and TCGA cohorts.
In the KCCH and LTHT cohort, KRAS gene copy number was determined by multiplex ligation-dependent probe amplification (MLPA) using the Salsa-FAM-labeled MLPA reagent kit and probemix P458-A1 or the updated version -B1 (MRC Holland, Amsterdam, The Netherlands) as previously described [25]. For further details on the KRAS probes included in this probemix see Supplementary Table 1. Fragment analysis of the MLPA reaction product was performed using capillary electrophoresis ABI-3130 XL (Applied Biosystems, California, USA) as previously described [25]. Failed experiments were repeated at least twice before a case was finally excluded from the analyses.
KRAS DNA copy number data from 237 KCCH GC has been previously published [25], but was re-analysed using a different methodology in the current study. The output files (FSA files) from the sequencer were initially imported into Coffalyser.net for fragment analysis and results were exported as csv files. Subsequent analyses were performed using the MLPAInter method, as previously described [26], implemented in R. Samples were normalised per batch using reference samples processed in each batch. Quality control was performed to exclude samples with low overall intensity, with a large difference in intensity between short probes and long probes, with low intensity of denaturation controls, or high within gene variation, defined as the average of the standard deviation of log-transformed values. Final values were calculated by averaging the peak height of each probe and then averaging the results of replicates. Copy number thresholds were set based on previously published studies [25, 27, 28], with a DNA copy number > 1.31 categorised as amplification. This analysis was performed separately for KCCH and LTHT cohorts.
In TCGA, KRASamp were determined by array-based somatic copy number analysis [29]. Level 3 copy number segmentation data was downloaded from the TCGA data portal [19] and used to estimate copy number for KRAS. Based on previous studies, a LogRatio > 0.4 was categorised as amplification [30].
KRAS mutation status
KRASmut data from a previous study were available for 230 KCCH and 275 LTHT GC patients [17]. KRASmut testing was performed on an additional 12 KCCH GCs as previously described [17]. In TCGA, KRASmut status was determined by whole-exome sequencing [29] and results were downloaded from the TCGA database portal [19] for 289 patients. In the TMGH cohort, KRAS (codon 12 and 13) was examined by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP), using primers and methods previously described [31, 32].
Microsatellite instability (MSI) status
Immunohistochemistry of DNA mismatch repair proteins were used as a surrogate marker of MSI status. Results for MLH1, MSH2, MSH6 and PMS2 were available from 230 KCCH GCs, and MLH1 and MSH2 from 253 LTHT GCs from a previous study [17]. MLH1, MSH2, MSH6 and PMS2 immunohistochemistry was performed on additional 13 GCs from the KCCH cohort for this study, as previously described [17].
In TCGA, MSI was determined by a DNA based MSI-Mono-Derived-Dinucleotide Assay using four mononucleotide repeat loci and three dinucleotide repeat loci using a multiplex fluorescent-labeled PCR and capillary electrophoresis [29]. Results were obtained from the TCGA database portal [19] for 295 GC patients. MSI-low GCs were grouped with microsatellite stable (MSS) GCs for further analyses following current guidelines [33].
In the TMGH cohort, mononucleotide repeats BAT25 and BAT26 were investigated, as previously described [34,35,36] and GC were classified as MSS or MSI.
Statistical analyses
All statistical analyses were performed using SPSS software version 23 (SPSS Inc., Chicago, III). The relationship between KRASmut or KRASamp and clinicopathological variables (age, gender, depth of invasion (pT), lymph node status (pN), TNM stage, Lauren classification [22], JGCA classification [21], MSI status and morphological heterogeneity status) was assessed using Chi-squared or Fisher’s exact test. The relationship between KRASmut and survival in LTHT and KCCH cohorts has been published previously [17]. Combining all cohorts, the relationship between KRASmut or KRASamp and 5-year overall survival was analysed using the Kaplan–Meier method and differences were assessed using the log rank test. A p value of < 0.05 was considered significant.
Results
Patient characteristics
The median (range) age of GC patients was as follows; KCCH: 65 years (35–85 years), LTHT: 72 years (14–96 years), TCGA: 68 years (35–90 years), TMGH: 78 years (51–96 years). For a summary of other patient clinicopathological variables in each cohort see Table 2.
Histological classification of gastric cancer
Histological classification was available for 1271 GCs. Using the JGCA classification, the most predominant phenotype was moderately differentiated tubular [tub2] (n = 408, 32%), followed by poorly differentiated solid type [por1] (n = 229, 18%), poorly differentiated non-solid type [por2] (n = 227, 18%), well-differentiated tubular [tub1] (n = 219, 17%), papillary [pap] (n = 71, 6%), signet-ring cell [sig] (n = 66, 5%) and mucinous [muc] (n = 51, 4%). According to Lauren classification, 293 (23%) GCs were classified as diffuse type, 698 (55%) as intestinal type, 51 (4%) as mucinous and 229 (18%) as indeterminate. Seven hundred and twenty-four GCs (57%) had intratumour morphological heterogeneity (see Table 2).
KRAS mutation status and relationship with clinicopathological variables
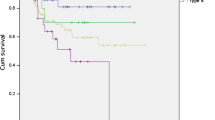
KRASmut status was available from 1266 GCs (KCCH n = 242; LTHT n = 275; TCGA n = 289, TMGH n = 460). In total, 68 (5%) GCs were KRAS mutant, with the highest frequency of KRASmut in the TCGA cohort (10%) and lowest frequency in the TMGH cohort (3%), see Table 2. Within the KRASmut GC, the most frequent histological phenotype was intestinal type (n = 41, 61%) or tub2 (n = 27, 40%) by Lauren and JGCA classification, respectively (see Fig. 1a). Comparing individual histological subtypes, mucinous phenotype displayed the highest frequency of KRASmut by Lauren (p = 0.013) and JGCA (p = 0.012) classification, respectively (see Fig. 1b). KRASmut was more frequent in MSI GC (p < 0.001). For the comparison of KRASmut status and other clinicopathological variables, see Table 3. The 5-year overall survival rate in patients with KRASmut or KRAS wild type GC was 63.6% and 54.8%, respectively, p = 0.541, see Fig. 2a.

Example of KRAS mutated GC with a moderately differentiated tubular (tub2) phenotype and b mucinous (muc) phenotype

Kaplan–Meier plots showing probability of overall survival in GC patients stratified by KRAS gene activation status. a Kaplan–Meier survival analysis showed no difference in survival when patients were stratified by KRAS mutation status. b Kaplan–Meier survival analysis showed no difference in survival when patients were stratified by KRAS amplification status
KRAS amplification and relationship with clinicopathological variables
KRAS gene copy number status was available from 649 GCs (KCCH n = 208, LTHT n = 216, TCGA n = 225). In total, 47 (7%) GCs had a KRASamp [TCGA (8%), LTHT (8%) and KCCH (6%)], see Table 2. Within KRASamp GC, intestinal type (n = 23, 50% or tub2 (n = 21, 46%) was the most frequent histological phenotype by Lauren and JGCA classification, respectively (see Fig. 3a). Comparing individual histological subtypes, KRASamp was more frequently found in indeterminate type (n = 12, 10%) or por1 (n = 12, 10%) phenotype by Lauren and JGCA classification, respectively (see Fig. 3b). There was no relationship between KRASamp and histological phenotype or any other clinicopathological variables, see Table 4. The 5-year overall survival rate in GC patients with and without KRASamp was 47.6% versus 55.6%, respectively, p = 0.166, see Fig. 2b.

Example of KRAS amplified GC with a moderately differentiated tubular (tub2) adenocarcinoma and b poorly differentiated solid-type (por1) adenocarcinoma
Only two GCs from the TCGA cohort had a concurrent KRASamp and KRASmut; one was a mucinous GC, the other was a por2 GC according to JGCA classification.
Discussion
This is the largest multicentre study to date to investigate the relationship between KRAS activation by mutation and/or amplification and histological phenotype in GC. The frequency of KRASamp (7%) was slightly higher than that of KRASmut (5%) which is consistent with other GC studies [10, 11, 37]. The higher frequency of KRASmut in the TCGA GC cohort compared to the other cohorts could be related to the methodology as TCGA used whole-exome sequencing to test non-hotspot regions, whereas other studies used less-sensitive Sanger sequencing/PCR–RFLP. We found KRASamp and KRASmut were exclusive in > 99% of GC, which is consistent with previous reports [11,12,13, 38].
The relationship between KRASmut and histological phenotype has not been investigated in great detail and previous studies were limited by small sample sizes and hence lack of statistical power [6]. In our study, we identified a relationship between KRASmut and mucinous histological phenotype, which is concordant with higher frequencies of KRASmut being reported in mucinous lung [8], ovarian [9] and colorectal cancer [39, 40]. However, due to the relatively low frequency of GC with mucinous phenotype and KRASmut (12%), it would not be feasible to use the presence of a mucinous phenotype as a predictor for the presence of a KRASmut in GC. The main component of mucinous GCs is extracellular mucin, which consists of high molecular weight glycoproteins regulated by expression of the MUC2, MUC5AC and MUC6 genes in humans [41]. In mouse models with constitutively activated KRAS in the stomach, irregular MUC4+ cells were found with abnormal mucins confirmed by Alcian-blue staining [42]. Interestingly, our study suggests a relationship between KRASmut and mucinous phenotype, which is characterised by extracellular mucin, but is not related to signet-ring cell type GC, which is characterised by intracellular mucin. Our study confirmed the relationship between KRASmut and the presence of MSI, which our group and others have described previously in a smaller GC cohort [43, 44].
The prognostic significance of KRASmut in GC remains controversial [6]. In our study, there was no association with the presence of KRASmut and survival. Interestingly, in lung and colorectal cancer, KRASmut has been associated with a poor prognosis [45, 46], whereas in ovarian cancer, KRASmut has been associated with an improved prognosis [47].
The relationship between KRASamp and clinicopathological variables, including histological phenotype in cancer is not well studied. In GC, we found no statistically significant relationship between KRASamp and histological phenotype, or any other clinicopathological variables. In contrast, others found that the presence of KRASamp is associated with a poor prognosis in GC [3, 10, 12]. This difference might be due to case selection and methodology used.
In our study, we used the JGCA scheme for the histological classification of GC and performed a conversion to the Lauren scheme, which is the most widely used histological classification system in Western countries [22]. Previous studies investigating the relationship between KRASmut and histological phenotype performed classification according to the Lauren scheme [6], for which there is no separate category for mucinous GC. The relatively large number of GCs classified as indeterminate according to the Lauren scheme comes from conversion from the JGCA por1 histological phenotype. Direct classification according to the Lauren scheme, would likely result in a higher proportion of GCs classified as either intestinal or diffuse.
In colorectal cancer, KRASmut is known to be an early event in the progression from normal colonic epithelial cell to adenoma, and finally to carcinoma [48]. The evidence of sequential development by accumulation of genetic alterations, including KRASmut, is still controversial in GC [49,50,51]. We were unable to make any comments regarding the role of KRAS activation in gastric carcinogenesis in our cohort as we did not investigate precancerous lesions in the current study. However, evidence from mouse models suggest that KRASmut is one of the key molecular alterations involved in the development of stomach dysplasia [52] and GC [53]. Based on the evidence from other cancer types that KRASmut influence the progression of a mucinous histological phenotype, we therefore speculate based on our results, that KRASmut in GC is an early event in GC development, whereas KRASamp is likely to be a late event occurring after the histological phenotype has been established. This would correspond with experiments in mice expressing oncogenic KRAS in combination with E-cadherin and p53 loss, which resulted in a rapid progression of GC compared to wild type mice [53].
Our study has some limitations. This is a retrospective study. Histological phenotyping was performed on a single slide. Given the high frequency of intra-tumoural morphological heterogeneity in this study and the previously reported intra-tumoural heterogeneity in KRASmut status in GC [54], the sensitivity of some of the techniques used in the current study may not be sufficient to detect KRAS activation in subclones of tumour cells. As we did not perform microdissection of tumour subregions, we cannot comment on KRAS status heterogeneity within the same tumour. Furthermore, we used different techniques for DNA extraction, KRASmut status analysis and MSI analysis in different cohorts included in the current study, each with differing sensitivities [55, 56].
In summary, we identified a relationship between KRASmut and mucinous histological phenotype in GC. The high level of intratumour morphological heterogeneity could reflect KRASmut heterogeneity, which may explain the failure of anti-EGFR therapy in GC.
Change history
06 June 2019
In the original publication of this article, Fig. 2 was published incorrectly. The correct Fig. 2 is given in this correction.
References
Gullo I, Carneiro F, Oliveira C, Almeida GM. Heterogeneity in gastric cancer: from pure morphology to molecular classifications. Pathobiology. 2018;85(1–2):50–63.
Dias Carvalho P, Guimaraes CF, Cardoso AP, Mendonca S, Costa AM, Oliveira MJ, et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. 2018;78(1):7–14.
Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24(7):968–77.
Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960.
Singh H, Longo DL, Chabner BA. Improving prospects for targeting RAS. J Clin Oncol. 2015;33(31):3650–9.
Hewitt LC, Hutchins GG, Melotte V, Saito Y, Grabsch HI. KRAS, BRAF and gastric cancer. Transl Gastrointest Cancer. 2015;4(6):429–47.
Er TK, Chen CC, Bujanda L, Herreros-Villanueva M. Current approaches for predicting a lack of response to anti-EGFR therapy in KRAS wild-type patients. BioMed Res Int. 2014;2014:591867.
Cha YJ, Shim HS. Biology of invasive mucinous adenocarcinoma of the lung. Transl Lung Cancer Res. 2017;6(5):508–12.
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384(9951):1376–88.
Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–84.
Mita H, Toyota M, Aoki F, Akashi H, Maruyama R, Sasaki Y, et al. A novel method, digital genome scanning detects KRAS gene amplification in gastric cancers: involvement of overexpressed wild-type KRAS in downstream signaling and cancer cell growth. BMC Cancer. 2009;9:198.
Qian Z, Zhu G, Tang L, Wang M, Zhang L, Fu J, et al. Whole genome gene copy number profiling of gastric cancer identifies PAK1 and KRAS gene amplification as therapy targets. Genes Chromosomes Cancer. 2014;53(11):883–94.
Ali SM, Sanford EM, Klempner SJ, Rubinson DA, Wang K, Palma NA, et al. Prospective comprehensive genomic profiling of advanced gastric carcinoma cases reveals frequent clinically relevant genomic alterations and new routes for targeted therapies. Oncologist. 2015;20(5):499–507.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21(5):449–56.
Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer. 2015;18(1):65–766.
Ooi A, Oyama T, Nakamura R, Tajiri R, Ikeda H, Fushida S, et al. Gene amplification of CCNE1, CCND1, and CDK6 in gastric cancers detected by multiplex ligation-dependent probe amplification and fluorescence in situ hybridization. Hum Pathol. 2017;61:58–67.
van Grieken NC, Aoyma T, Chambers PA, Bottomley D, Ward LC, Inam I, et al. KRAS and BRAF mutations are rare and related to DNA mismatch repair deficiency in gastric cancer from the East and the West: results from a large international multicentre study. Br J Cancer. 2013;108(7):1495–501.
Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo D, et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–677.
The Cancer Genome Atlas. Comprehensive molecular characterization of gastric adenocarcinoma. National Institutes of Health; 2014 . https://tcga-data.nci.nih.gov/docs/publications/stad_2014/. Accessed 2 July 2016
Washington K. 7th edition of the AJCC cancer staging manual: stomach. Ann Surg Oncol. 2010;17(12):3077–9.
Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer. 2011;14(2):101–12.
Lauren P. The two histological main types of gastric carcinoma: diffuse and so called intestinal-type carcinoma. Acta Pathol Microbiol Scand. 1965;64:31–49.
Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29(10):1261–70.
Arai T, Matsuda Y, Aida J, Takubo K, Ishiwata T. Solid-type poorly differentiated adenocarcinoma of the stomach: clinicopathological and molecular characteristics and histogenesis. Gastric Cancer. 2019;22(2):314–22.
Silva AN, Coffa J, Menon V, Hewitt LC, Das K, Miyagi Y, et al. Frequent coamplification of receptor tyrosine kinase and downstream signaling genes in Japanese primary gastric cancer and conversion in matched lymph node metastasis. Ann Surg. 2016;267(1):114–21.
van Eijk R, Eilers PH, Natte R, Cleton-Jansen AM, Morreau H, van Wezel T, et al. MLPAinter for MLPA interpretation: an integrated approach for the analysis, visualisation and data management of Multiplex Ligation-dependent Probe Amplification. BMC Bioinform. 2010;11:67.
Ooi A, Inokuchi M, Harada S, Inazawa J, Tajiri R, Kitamura SS, et al. Gene amplification of ESR1 in breast cancers—fact or fiction? A fluorescence in situ hybridization and multiplex ligation-dependent probe amplification study. J Pathol. 2012;227(1):8–16.
Bunyan DJ, Eccles DM, Sillibourne J, Wilkins E, Thomas NS, Shea-Simonds J, et al. Dosage analysis of cancer predisposition genes by multiplex ligation-dependent probe amplification. Br J Cancer. 2004;91(6):1155–9.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Hanemaaijer NM, Sikkema-Raddatz B, van der Vries G, Dijkhuizen T, Hordijk R, van Essen AJ, et al. Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet. 2012;20(2):161–5.
Ohshima S, Shimizu Y, Takahama M. Detection of c-Ki-ras gene mutation in paraffin sections of adenocarcinoma and atypical bronchioloalveolar cell hyperplasia of human lung. Virchows Arch. 1994;424(2):129–34.
Suzuki Y, Orita M, Shiraishi M, Hayashi K, Sekiya T. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene. 1990;5(7):1037–43.
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–8.
Zhou XP, Hoang JM, Cottu P, Thomas G, Hamelin R. Allelic profiles of mononucleotide repeat microsatellites in control individuals and in colorectal tumors with and without replication errors. Oncogene. 1997;15(14):1713–8.
Cravo M, Lage P, Albuquerque C, Chaves P, Claro I, Gomes T, et al. BAT-26 identifies sporadic colorectal cancers with mutator phenotype: a correlative study with clinico-pathological features and mutations in mismatch repair genes. J Pathol. 1999;188(3):252–7.
Esemuede I, Forslund A, Khan SA, Qin LX, Gimbel MI, Nash GM, et al. Improved testing for microsatellite instability in colorectal cancer using a simplified 3-marker assay. Ann Surg Oncol. 2010;17(12):3370–8.
Warneke VS, Behrens HM, Haag J, Balschun K, Boger C, Becker T, et al. Prognostic and putative predictive biomarkers of gastric cancer for personalized medicine. Diagn Mol Pathol. 2013;22(3):127–37.
Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013;133(5):1259–65.
Rosty C, Young JP, Walsh MD, Clendenning M, Walters RJ, Pearson S, et al. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol. 2013;26(6):825–34.
Khan M, Loree JM, Advani SM, Ning J, Li W, Pereira AAL, et al. Prognostic implications of mucinous differentiation in metastatic colorectal carcinoma can be explained by distinct molecular and clinicopathologic characteristics. Clin Colorectal Cancer. 2018;17(4):E699–E709.
Boltin D, Niv Y. Mucins in gastric cancer—an update. J Gastrointest Dig Syst. 2013;3(123):15519.
Kinoshita H, Hayakawa Y, Konishi M, Hata M, Tsuboi M, Hayata Y, et al. Three types of metaplasia model through Kras activation, Pten deletion, or Cdh1 deletion in the gastric epithelium. J Pathol. 2019;247(1):35–47.
Polom K, Das K, Marrelli D, Roviello G, Pascale V, Voglino C, et al. KRAS mutation in gastric cancer and prognostication associated with microsatellite instability status. Pathol Oncol Res. 2019;25(1):333–40.
Zhao W, Chan TL, Chu KM, Chan AS, Stratton MR, Yuen ST, et al. Mutations of BRAF and KRAS in gastric cancer and their association with microsatellite instability. Int J Cancer. 2004;108(1):167–9.
Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90(9):675–84.
Zhang J, Park D, Shin DM, Deng X. Targeting KRAS-mutant non-small cell lung cancer: challenges and opportunities. Acta Biochim Biophys Sin (Shanghai). 2016;48(1):11–6.
Nodin B, Zendehrokh N, Sundstrom M, Jirstrom K. Clinicopathological correlates and prognostic significance of KRAS mutation status in a pooled prospective cohort of epithelial ovarian cancer. Diagn Pathol. 2013;8:106.
Arvelo F, Sojo F, Cotte C. Biology of colorectal cancer. Ecancermedicalscience. 2015;9:520.
Maesawa C, Tamura G, Suzuki Y, Ogasawara S, Sakata K, Kashiwaba M, et al. The sequential accumulation of genetic alterations characteristic of the colorectal adenoma-carcinoma sequence does not occur between gastric adenoma and adenocarcinoma. J Pathol. 1995;176(3):249–58.
Gong C, Mera R, Bravo JC, Ruiz B, Diaz-Escamilla R, Fontham ET, et al. KRAS mutations predict progression of preneoplastic gastric lesions. Cancer Epidemiol Biomark Prev. 1999;8(2):167–71.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–54.
Okumura T, Ericksen RE, Takaishi S, Wang SSW, Dubeykovskiy Z, Shibata W, et al. K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res. 2010;70(21):8435–45.
Till JE, Yoon C, Kim BJ, Roby K, Addai P, Jonokuchi E, et al. Oncogenic KRAS and p53 loss drive gastric tumorigenesis in mice that can be attenuated by E-cadherin expression. Cancer Res. 2017;77(19):5349–59.
Queiros P, Pinheiro H, Carvalho J, Oliveira P, Gullo I, Carneiro F, et al. KRAS mutations in microsatellite instable gastric tumours: impact of targeted treatment and intratumoural heterogeneity. Virchows Arch. 2015;467(4):383–92.
Matsunaga M, Kaneta T, Miwa K, Ichikawa W, Fujita KI, Nagashima F, et al. A comparison of four methods for detecting KRAS mutations in formalin-fixed specimens from metastatic colorectal cancer patients. Oncol Lett. 2016;12(1):150–6.
de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol. 2010;28(20):3380–7.
Acknowledgements
The authors thank S. Kanari for technical assistance.
Funding
This study was supported in part by the JSPS KAKENHI Grant numbers JP25460428 and JP16K08664 (TA). HG has received funding from The Pathological Society of Great Britain and Ireland, Yorkshire Cancer Research and Sasakawa Foundation UK. TY has received funding from Sasakawa Foundation UK and Kanagawa Standard Anti-cancer Therapy Support System (Japan).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hewitt, L.C., Saito, Y., Wang, T. et al. KRAS status is related to histological phenotype in gastric cancer: results from a large multicentre study. Gastric Cancer 22, 1193–1203 (2019). https://doi.org/10.1007/s10120-019-00972-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-00972-6