
Abstract
Background
Ramucirumab, a monoclonal antibody vascular endothelial growth factor receptor-2 antagonist, given as monotherapy improved survival in a global phase 3 study (REGARD) of patients with gastric cancer. However, REGARD did not include Japanese patients. This study evaluated the efficacy and safety of ramucirumab monotherapy in Japanese patients with advanced gastric cancer.
Methods
This multicenter, open-label, nonrandomized phase 2 study (Clinicaltrials.gov: NCT01983878) was performed at 16 Japanese sites. Patients with advanced gastric or gastroesophageal junction cancer after disease progression following first-line chemotherapy received intravenous ramucirumab 8 mg/kg every 2 weeks. Primary efficacy outcome: 12-week progression-free survival rate (PFS).
Results
Thirty-six patients were enrolled. The 12-week PFS rate was 23.8% [90% confidence interval (CI) 12.4–37.2); the primary outcome was not met as the lower limit of the CI was outside the threshold of 16%. Median PFS was 6.6 weeks (90% CI 6.1–7.1). No patients achieved an objective response, and 11 (31%) patients achieved disease control. Median overall survival was 8.6 months (90% CI 5.7–10.7). The most frequent treatment-emergent adverse events (TEAEs) were diarrhea (9/36; 25%) and decreased appetite (8/36; 22%). Three patients reported Grade ≥ 3 ileus; all other Grade ≥ 3 TEAEs were reported by ≤ 2 patients. The most frequent adverse events of special interest (AESIs) were hypertension (10/36; 28%), bleeding/hemorrhage (7/36; 19%), and proteinuria (7/36; 19%). All Grade ≥ 3 AESIs were reported by ≤ 2 patients.
Conclusions
These findings suggest that ramucirumab monotherapy has clinical activity and a manageable safety profile in Japanese patients with gastric cancer after disease progression following first-line chemotherapy.
Similar content being viewed by others


Introduction
Gastric cancer is the most common type of cancer in Japan and is associated with a poor prognosis. Of note, the incidence and age-adjusted incidence of gastric cancer in Japan in 2012 were 107,898 and 29.9 per 100,000, respectively [1]. In addition, gastric cancer was the second leading cause of cancer-related death in Japan in 2012, accounting for 52,326 deaths [1]. This high mortality rate reflects the advanced disease presentation of most patients [2] and the lack of effective treatment options. The current standard of care for first-line treatment of advanced/metastatic gastric cancer is systemic chemotherapy, specifically the combination of fluoropyrimidine- and platinum-based agents [3]. Although second-line treatment has been recently established, there is a need for additional therapeutic options.
Ramucirumab is a monoclonal antibody vascular endothelial growth factor (VEGF) receptor-2 (VEGFR-2) antagonist that has demonstrated efficacy in the treatment of advanced gastric cancer. Specifically, two multinational studies, REGARD [4] and RAINBOW [5], have shown that ramucirumab monotherapy or combination therapy with paclitaxel improves survival compared with placebo or placebo plus paclitaxel, respectively, in patients with advanced gastric or gastroesophageal junction cancer after failed first-line chemotherapy. Ramucirumab was subsequently approved in Japan for unresectable, advanced/recurrent gastric cancer in 2015 [6] and ramucirumab combined with weekly paclitaxel and ramucirumab monotherapy were listed in 4th edition of Japanese treatment guidelines for gastric cancer [7].
A recent subgroup analysis of the RAINBOW study demonstrated that the safety profile of ramucirumab combined with paclitaxel was similar in Japanese and Caucasian patients and that progression-free survival (PFS) and the objective response rate (ORR) were improved relative to placebo plus paclitaxel in both populations [8]. Given that survival outcomes can differ between Asian and Caucasian patients with gastric cancer [9], additional information on the efficacy and safety of ramucirumab monotherapy in Japanese patients with advanced gastric cancer is eagerly awaited.
The aim of this phase 2 study was to evaluate the efficacy, safety, and pharmacokinetics of ramucirumab monotherapy in Japanese patients with advanced gastric or gastroesophageal junction cancer after disease progression following first-line chemotherapy.
Methods
Study design
This multicenter, open-label, nonrandomized phase 2 study was carried out at 16 sites in Japan and involved patients with metastatic or locally recurrent gastric or gastroesophageal junction adenocarcinoma who had disease progression after one prior chemotherapy.
The study protocol was approved by applicable ethical review boards and the study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and applicable regulations. The study was registered at http://www.clinicaltrials.gov (NCT01983878).
All patients provided written informed consent before study entry.
Study population
Patients with histologically or cytologically confirmed metastatic or locally recurrent unresectable gastric or gastroesophageal junction adenocarcinoma and disease progression on prior chemotherapy were eligible for inclusion. The main inclusion criteria were: age ≥ 20 years; measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST), Version 1.1, and/or evaluable disease; disease progression during or within 4 months after the last dose of first-line therapy for metastatic disease or during or within 6 months after the last dose of adjuvant therapy; life expectancy ≥ 12 weeks; Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1; adequate recovery from toxicities/effects of prior therapy; and adequate hematologic, hepatic, coagulation, and renal function. The main exclusion criteria were: documented and/or symptomatic brain or leptomeningeal metastases; any Grade 3 or 4 gastrointestinal bleeding within 3 months before enrollment; any arterial thromboembolic event within 6 months before enrollment; ongoing or active significant infection, symptomatic congestive heart failure, unstable angina pectoris, symptomatic or poorly controlled cardiac arrhythmia, uncontrolled thromboembolic or hemorrhagic disorder, or any other serious uncontrolled medical disorders in the opinion of the investigators; poorly controlled hypertension, despite standard medical management; serious or nonhealing wound, ulcer, or bone fracture within 28 days before enrollment; prior therapy with any direct inhibitor of VEGF or VEGFR-2 (including bevacizumab) or any antiangiogenic agent; and current chronic antiplatelet therapy, including aspirin [once-daily aspirin use (maximum dose 325 mg/day) was permitted], nonsteroidal anti-inflammatory drugs dipyridamole or clopidogrel, or similar agents.
Treatment protocol
Patients received ramucirumab 8 mg/kg intravenously over approximately 60 min once every 2 weeks (1 cycle) with radiographic assessments of disease status every 6 weeks. Treatment was continued until there was evidence of progressive disease, unacceptable toxicity, withdrawal of consent, or until other withdrawal criteria were met.
Efficacy outcome measures
The primary efficacy outcome was the 12-week PFS rate, defined as the probability of being alive and progression-free at 12 weeks after study enrollment.
Secondary efficacy outcomes included PFS, ORR, disease control rate (DCR), and overall survival (OS). Progression-free survival was defined as the time from study enrollment to the first observation of objective progression or death from any cause. The ORR was defined as the proportion of patients who achieved a best overall response of complete response (CR) or partial response (PR). The DCR was defined as the proportion of patients who achieved a best overall response of CR, PR, or stable disease (SD). Overall survival was defined as the time from study enrollment to death from any cause.
Assessments of response, according to RECIST, Version 1.1, were carried out every 6 weeks, with confirmatory assessment for patients with an objective response of PR or CR obtained at least 4 weeks later. Survival measurements were carried out every 3 months. An Independent Radiography Review Committee (IRRC) independently reviewed computed tomography scans and magnetic resonance imaging data for tumor assessments.
Pharmacokinetics
Blood samples were collected from 6 patients immediately before, 1 h after start of the infusion, and at 1, 4, 23, 47, 95, 167, 263, and 335 h after the completion of the ramucirumab infusion (Day 1 of Cycle 1). Pharmacokinetic parameter estimates for ramucirumab were calculated by standard noncompartmental analysis. Blood samples were also collected from all patients before each cycle for calculation of the minimum blood plasma concentration (Cmin).
Pharmacokinetic analyses were performed using Phoenix WinNonlin Professional 6.3 (Certara, Princeton, USA). Exposure–response analyses were performed using S-PLUS 8.2 for Windows (TIBCO Software Inc, Palo Alto, USA).
Safety
All patients were assessed regularly for treatment-emergent adverse events (TEAEs) from the time of first infusion to 30 days after the last infusion of ramucirumab. Clinical laboratory assessments were carried out at regular intervals during the study.
Immunogenicity
Blood samples were collected before each treatment cycle and at the 30-day safety follow-up visit for analysis of anti-ramucirumab antibodies.
Statistical analysis
A sample size of 27 was required based on the expected and threshold 12-week PFS rate of 40 and 16%, respectively, a 1-sided significance level of 0.05, and desired power of 90%. To allow for censoring of some cases, the target number of patients for enrollment was 33.
The efficacy and safety analysis populations included patients who received at least 1 dose of the study drug [Full Analysis Set (FAS)]. The pharmacokinetic analysis population included a subset of patients who underwent intensive sampling to estimate pharmacokinetic parameters and all patients with available ramucirumab concentration data before ramucirumab infusion for calculation of Cmin.
Tumor evaluations by the IRRC were used for the primary analysis of the 12-week PFS rate and the secondary outcomes of PFS, ORR, and DCR. Tumor evaluations by the investigators were used for sensitivity analysis of the 12-week PFS rate. The 12-week PFS rate was estimated using the Kaplan–Meier method with PFS time as a time-to-event variable. The median estimate and two-sided 90% confidence interval (CI; calculated using log–log transformed survival function) of the PFS rate were determined. Kaplan–Meier curves of PFS and OS were generated, along with the median time and 90% CI. The ORR and DCR with 90% CIs were summarized.
Treatment-emergent adverse events were summarized (using descriptive statistics) by Medical Dictionary for Regulatory Activities System Organ Class and preferred term. Severity was summarized according to the most severe National Cancer Institute—Common Terminology Criteria for Adverse Events, Version 4.03, grade as assigned by the investigators.
Analyses were performed using SAS Version 9.2 (SAS Institute, Cary, USA) or later.
Results
Disposition
A total of 36 patients were enrolled, all of whom received at least 1 dose of ramucirumab. All patients discontinued study treatment due to PD (32/36; 89%) or adverse events (4/36; 11%).
Demographic and baseline clinical characteristics
The majority (> 50%) of patients were male, had an ECOG PS of 0, and had the primary tumor located in the stomach body (Table 1). Intestinal and diffuse were the most common histological subtypes, accounting for > 75% of patients. All patients had received prior systemic therapy and most (69%) had prior surgery.
Efficacy outcomes
Primary
The IRRC-evaluated 12-week PFS rate was 23.8% (90% CI: 12.4, 37.2). As the lower limit of the 90% CI for the 12-week PFS rate was outside the predetermined threshold (16%), the primary outcome was not met. The investigator-evaluated 12-week PFS rate was 31.4% (90% CI: 19.2, 44.4).
Secondary
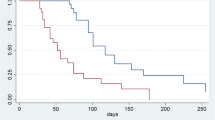
The median PFS time was 6.6 weeks (90% CI: 6.1, 7.1) (Fig. 1). No patients achieved an objective response, whereas 11 (31%) patients achieved disease control (Table 2). The median OS time was 8.6 months (90% CI: 5.7, 10.7) (Fig. 2).

Kaplan–Meier curve of progression-free survival. CI confidence interval, PFS progression-free survival

Kaplan–Meier curve of overall survival. CI confidence interval
Ramucirumab pharmacokinetics/pharmacodynamics
After a single intravenous dose (8 mg/kg), the half-life (t1/2) was 183 h [approximately, 8 days (Fig. 3)], the geometric mean clearance was 0.0150 L/h, and the volume of distribution at steady state was 3.29 L.

Serum concentration–time profile of ramucirumab following a single 8 mg/kg ramucirumab administration as a 60-min infusion on Day 1, Cycle 1. Data are presented as arithmetic mean ± standard deviation
After multiple (or repeated) intravenous doses (8 mg/kg every other week cycle), the geometric mean predose concentration for Dose 4 (Cmin after Dose 3; 67.8 μg/mL) was approximately twice that of the predose concentration for Dose 2 (Cmin after Dose 1; 32.5 μg/mL. The predose concentration from Dose 4 to Dose 6 showed little change (Fig. 4), indicating that steady state was achieved following 6 weeks of ramucirumab treatment.

Ramucirumab trough (prior to infusion) concentrations following administration of 8 mg/kg of ramucirumab every 2 weeks as an intravenous infusion over approximately 1 h. Outline of box shows the 25th and 75th percentile with inside lines being the arithmetic mean (solid) and median (dashed). Whiskers show the lowest and highest values within 1.5 times the difference between the 1st and 3rd quartile. Individual observations outside the whiskers are shown with open circles
There were no obvious relationships between exposure (Cmin after Dose 1) and treatment response. Median PFS time was 1.48 months in both the low (Cmin < median after Dose 1) and high (Cmin ≥ median after Dose 1) exposure groups; median OS time was 7.96 and 7.56 months in the low- and high-exposure groups, respectively.
Exposure and postdiscontinuation anticancer therapy
The median duration of therapy was 6 weeks (range 2–44 weeks) and the median relative dose intensity was 99.6% (range 88.5–107.8%).
After discontinuation, 24 (67%) patients received systemic anticancer therapy, most commonly paclitaxel (17/36; 47%), irinotecan hydrochloride (10/36; 28%), irinotecan (4/36; 11%), trastuzumab (4/36; 11%), and/or oxaliplatin (3/36; 8%).
Safety
All patients reported at least 1 TEAE and half of all patients reported at least 1 adverse event of special interest (AESI) (Table 3). The most frequently reported TEAEs (> 20% of patients) were diarrhea and decreased appetite. Three patients reported Grade ≥ 3 ileus; all other Grade ≥ 3 TEAEs were reported by 2 or fewer patients. The most frequently reported AESIs (> 19% of patients) were hypertension, bleeding/hemorrhage, and proteinuria. All Grade ≥ 3 AESIs were reported by 2 or fewer patients. No patients reported infusion-related/hypersensitivity reactions, gastrointestinal perforation, congestive heart failure, wound healing complication, fistula formation, reversible posterior leukoencephalopathy syndrome, or interstitial lung disease.
Three patients (8%) experienced TEAEs that led to discontinuation of treatment during the study period. The TEAEs (all Grade 3) were upper gastrointestinal hemorrhage, proteinuria, and hypertension (all n = 1).
No patients died while receiving ramucirumab; 1 patient died within 30 days of the last dose of ramucirumab due to study disease.
Analyses of clinical laboratory data did not reveal any new or increased safety concerns relative to the analysis of the adverse event data.
Immunogenicity
Anti-ramucirumab antibodies were detected in 1/36 (3%) patients. Neutralizing antibodies were not detected and the patient did not experience an infusion-related reaction.
Discussion
This is the first study to evaluate the efficacy, including 12-week PFS and response rate, safety, and pharmacokinetics of ramucirumab monotherapy in Japanese patients with advanced gastric or gastroesophageal junction cancer that had progressed after first-line chemotherapy. Although the primary outcome was not met, the estimate of 12-week PFS evaluated in 36 Japanese patients was consistent with clinical activity, and the safety and tolerability of ramucirumab were clinically manageable. These findings support the use of ramucirumab in the treatment of advanced gastric or gastroesophageal junction cancer after failed first-line chemotherapy.
The primary efficacy outcome of this study, 12-week PFS, supports the clinical activity of ramucirumab monotherapy in Japanese patients with gastric cancer. Specifically, the 12-week PFS rate (23.8%) was numerically higher than that for the placebo arm in REGARD (15.8%) [4]. Although the IRRC-evaluated 12-week PFS rate in this study seems lower than that for the analysis of the ramucirumab arm in REGARD (40.1%) and the lower limit of the 90% CI fell outside the predetermined threshold, the investigator-evaluated 12-week PFS rate (31.4%) in this study is similar to the REGARD finding, and is a more equitable comparator, given that PFS was assessed by the investigators in REGARD. Secondary efficacy outcomes were also supportive of clinical activity; median OS (8.6 months) and PFS [6.6 weeks (1.5 months)] were numerically longer than median OS and PFS for the placebo arm in REGARD (3.8 months and 1.3 months, respectively) [4], as was the disease control rate (31% in this study versus 23% in REGARD) [4]. The median length of OS is noteworthy and similar to the durations reported in the WJOG 4007 trial [10], which included Japanese patients with advanced gastric cancer who were treated with either paclitaxel (OS: 9.5 months) or irinotecan (OS: 8.4 months) after first-line failure of fluoropyrimidine plus platinum. No patients in this study achieved an objective response, consistent with the very low rates in REGARD (3% in both treatment arms) [4]. The findings from this study support the clinical activity of ramucirumab monotherapy in Japanese patients, also consistent with those of the subgroup analysis of Asian patients in REGARD, which demonstrated survival benefits (OS and PFS) with ramucirumab monotherapy [4].
The pharmacokinetics of ramucirumab in Japanese patients was generally similar to those reported in other studies of ramucirumab [11,12,13]. Of note, the half-life of ramucirumab in this Japanese population is similar to that determined in a predominantly Caucasian population of patients with advanced malignant solid tumors [11]. However, no exposure–response relationship was observed in this study, which contrasts with what has been observed in ramucirumab phase 3 trials [14]. Presumably, the sample size of this study is too small to draw conclusions about the consistency of the exposure–response phenomenon in this population.
The safety findings of this study are consistent with the known safety profile of ramucirumab. The majority of TEAEs in Japanese patients with gastric cancer were manageable and, as per REGARD [4], Grade ≥ 3 TEAEs were infrequent. Of note, toxicities related to cytotoxic chemotherapy, such as bone marrow toxicity and gastrointestinal symptoms, were relatively uncommon. Hypertension, a known side effect of antiangiogenic treatments, was the most common TEAE, although only 2 (6%) patients experienced Grade ≥ 3 hypertension. In REGARD, hypertension was more commonly reported in the ramucirumab arm than in the placebo arm (16 vs 8%) [4].
Consistent with the findings from other studies, including REGARD, in which patients received ramucirumab as monotherapy [4, 15, 16], a very small proportion (3%) of Japanese patients developed anti-ramucirumab antibodies in this study. This finding suggests that ramucirumab has a low immunogenicity in Japanese patients with gastric cancer.
The main strengths of this study include the prospective, multicenter design, and the homogenous Japanese population. Limitations include the small sample size and the lack of a control/comparator group.
The findings from this phase 2 study, the first clinical experience in a Japanese population, suggest that ramucirumab monotherapy has clinical activity and a manageable safety profile in Japanese patients with gastric cancer following disease progression after first-line platinum- or fluoropyrimidine-containing combination therapy. With reference to REGARD [4], which included a predominantly non-Asian patient population, there do not appear to be any marked differences in efficacy/safety of ramucirumab between Japanese and non-Japanese patients for this disease population. These findings support the clinical use of ramucirumab monotherapy in Japan for treating advanced gastric or gastroesophageal junction cancer after failed first-line chemotherapy. We await the results from future studies regarding potential predictive biomarkers for ramucirumab therapy.
References
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer incidence and mortality worldwide: IARC CancerBase No. 11. Lyon, France; International Agency for Research on Cancer. Available at http://globocan.iarc.fr. Accessed on 14 July 2016.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol. 2006;24:2903–9.
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–9.
Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–35.
Pharmaceuticals and Medical Devices Agency, Japan. List of approved products (2016). Available at http://www.pmda.go.jp/files/000212691.pdf. Accessed on 28 June 2016.
Japanese Gastric Cancer A. Japanese gastric cancer treatment guidelines 2014 (ver. 4). Gastric Cancer. 2017;20:1–19.
Shitara K, Muro K, Shimada Y, Hironaka S, Sugimoto N, Komatsu Y, et al. Subgroup analyses of the safety and efficacy of ramucirumab in Japanese and Western patients in RAINBOW: a randomized clinical trial in second-line treatment of gastric cancer. Gastric Cancer. 2016;19:927–38.
Howard JH, Hiles JM, Leung AM, Stern SL, Bilchik AJ. Race influences stage-specific survival in gastric cancer. Am Surg. 2015;81:259–67.
Hironaka S, Ueda S, Yasui H, Nishina T, Tsuda M, Tsumura T, et al. Randomized, open-label, phase III study comparing irinotecan with paclitaxel in patients with advanced gastric cancer without severe peritoneal metastasis after failure of prior combination chemotherapy using fluoropyrimidine plus platinum: WJOG 4007 trial. J Clin Oncol. 2013;31:4438–44.
Chow LQ, Smith DC, Tan AR, Denlinger CS, Wang D, Shepard DR, et al. Lack of pharmacokinetic drug-drug interaction between ramucirumab and paclitaxel in a phase II study of patients with advanced malignant solid tumors. Cancer Chemother Pharmacol. 2016;78:433–41.
Masuda N, Iwata H, Aogi K, Xu Y, Ibrahim A, Gao L, et al. Safety and pharmacokinetics of ramucirumab in combination with docetaxel in Japanese patients with locally advanced or metastatic breast cancer: a phase Ib study. Jpn J Clin Oncol. 2016;46(12):1088–94.
Wang D, Braiteh F, Lee JJ, Denlinger CS, Shepard DR, Chaudhary A, et al. Lack of pharmacokinetic drug-drug interaction between ramucirumab and irinotecan in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;78:727–33.
Tabernero J, Ohtsu A, Muro K, Van Cutsem E, Oh SC, Bodoky G, et al. Exposure-response (E-R) relationship of ramucirumab (RAM) from two global, randomized, double-blind, phase 3 studies of patients (Pts) with advanced second-line gastric cancer. J Clin Oncol. 2015;33(Suppl 3):121.
Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010;28:780–7.
Chiorean EG, Hurwitz HI, Cohen RB, Schwartz JD, Dalal RP, Fox FE, et al. Phase I study of every 2- or 3-week dosing of ramucirumab, a human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2 in patients with advanced solid tumors. Ann Oncol. 2015;26:1230–7.
Acknowledgements
The authors would like to thank all study participants.
Funding
This study was sponsored by Eli Lilly and Company, the manufacturer and licensee of CYRAMZA®. Medical writing assistance was provided by Luke Carey, PhD, and Justine Southby, PhD, CMPP, of ProScribe—Envision Pharma Group, and was funded by Eli Lilly and Company. ProScribe’s services complied with international guidelines for Good Publication Practice (GPP3).
Author information
Authors and Affiliations
Contributions
All authors participated in the interpretation of study results, and in the drafting, critical revision, and approval of the final version of the manuscript. TK, KM, and TS were involved in designing the study. KY, KF, FN, YO, NM, and TN were study investigators and involved in data collection. MT was involved in the statistical analysis. TK was involved in the pharmacokinetics analysis.
Corresponding author
Ethics declarations
Role of the sponsor
Eli Lilly and Company was involved in the study design, data collection, data analysis, and preparation of the manuscript.
Conflict of interest
KY and NM have received grants and personal fees from Eli Lilly and Company. FN has received grants from Taiho Pharmaceutical, Merck Serono, Zeria Pharmaceutical, Yakult, OncoTherapy Science, J-Pharma, Eli Lilly Japan K.K., Janssen Pharmaceutical, Bayer Yakuhin, Kyowa Hakko Kirin, Shionogi, Daiichi Sankyo, Takeda, Chugai Pharmaceutical, GlaxoSmithKline, Nippon Kayaku, Bristol-Myers Squibb, Mochida Pharmaceutical, Astellas Pharma, Ono Pharmaceutical, Sanofi S.A., Novartis, Torii Pharmaceutical, MSD, Covance Inc., Baxalta Japan, Eisai, NanoCarrier, and NPO Japan Clinical Research Support Unit. YO and TN have received grants from Eli Lilly and Company. TK, MT, and KM are employees of and own shares in Eli Lilly and Company. TS has received grants or personal fees from Yakult Honsha, Chugai Pharmaceutical, Ono Pharmaceutical, Eli Lilly and Company, Merck-Serono, Bristol-Myers Squibb, Takeda Pharmaceutical, Taiho Pharmaceutical, and Takara Bio. KF has no potential conflicts of interest to declare.
Human rights statement and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent or substitute for it was obtained from all patients for being included in the study.
Rights and permissions
About this article

Cite this article
Yamaguchi, K., Fujitani, K., Nagashima, F. et al. Ramucirumab for the treatment of metastatic gastric or gastroesophageal junction adenocarcinoma following disease progression on first-line platinum- or fluoropyrimidine-containing combination therapy in Japanese patients: a phase 2, open-label study. Gastric Cancer 21, 1041–1049 (2018). https://doi.org/10.1007/s10120-018-0811-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-018-0811-4