
Abstract
Background
Although genetic alterations in patients with advanced gastric cancer have been extensively studied, those in patients with intramucosal neoplasia (IMN) are still poorly understood.
Methods
We evaluated genetic differences in 158 IMNs, including 51 low-grade dysplasias, 58 high-grade dysplasias (HGDs), 30 intramucosal cancers (IMCs), and 19 mixed tumors (composed of IMC and HGD within the same tumor), using PCR-based microsatellite analysis [allelic imbalance (AI) and microsatellite instability (MSI)]. We classified the DNA methylation status as a hypermethylated epigenome, a moderately methylated epigenome, or a hypomethylated epigenome. In addition, p53 overexpression, β-catenin nuclear localization, and mucin expression were also examined.
Results
From cluster analysis, the IMNs examined were categorized into four subgroups as follows. Tumors in subgroup 1 were characterized by MSI-high status, a hypermethylated epigenome, and loss or reduction of expression of MLH-1. Tumors in subgroup 2 showed a mixed pattern consisting of AI and MSI. In contrast, tumors in subgroup 3, which showed accumulation of multiple AIs, were closely associated with HGD, IMC, or mixed tumor and exhibited nuclear expression of β-catenin. Tumors in subgroup 4, which were generally low-grade dysplasias, exhibited a low frequency of AIs and no MSI. Although the mucin phenotype was not correlated with any subgroup, expression of mucin was associated with some subgroups. Overexpression of p53 was common in all subgroups.
Conclusion
The approach described herein was useful for studying genetic differences in IMNs. In addition, we suggest that stratification of genetic differences may help to identify genetic molecular profiles in IMNs.
Similar content being viewed by others
Introduction
Despite a considerable decrease in the incidence of gastric cancer (GC) in many developed countries, GC remains the fourth most commonly diagnosed malignancy and the second leading cause of cancer-related death worldwide [1, 2]. In the past few decades, standard multimodal treatment strategies have been used to improve outcomes in patients with GC, particularly those with advanced and metastatic diseases [2]. Thus, evaluation of the molecular mechanisms underlying the development of early-stage GC is expected to improve the efficacy of therapeutic management strategies for patients with GC.
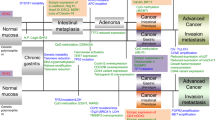
Gastric carcinogenesis is a multistep process involving genetic and epigenetic alterations in proto-oncogenes, tumor-suppressor genes, and mismatch-repair genes [3–5]. Previous studies have shown that there are two classical pathways associated with the development of colorectal cancers (CRCs) and GCs: microsatellite instability (MSI) and chromosomal instability (CIN) [6–10]. A recent study showed that molecular alterations in GC can be classified into four subgroups; that is, MSI-type tumors, CIN-type tumors, Epstein–Barr virus (EBV)-associated tumors, and genomically stable (GS) tumors [8]. The MSI phenotype (MSI-high tumors) is genetically characterized by MSI and genome-wide hypermethylation [11, 12]. Furthermore, the MSI phenotype shows characteristic histological features and favorable prognosis compared with microsatellite stable (MSS) cancer [7, 8]. On the other hand, CIN is closely associated with accumulation of multiple losses of heterozygosity, p53 mutations, and DNA aneuploidy (e.g., chromosomal damage in tumor cells) [6–8]. EBV-associated GC is a distinct subtype that accounts for nearly 10 % of GCs. It is defined by monoclonal expansion of cancer cells with latent EBV infection [8]. EBV-associated GC shows characteristic clinicopathological features, including predominance among men, a proximal location in the stomach, lace pattern and lymphoepithelioma-like histologic features, and a favorable prognosis [8]. In addition, although EBV-associated tumors are characterized by CpG island methylation phenotype (CIMP)-high status, MSI is not detected in this phenotype [3]. Finally, the GS phenotype, which is distinct from the CIN phenotype, is frequently observed in diffuse-type GC. GSSs are frequently detected in the diffuse histological variant and enriched for mutations in RHOA or fusions involving the Rho family of GTPase-activating proteins [3, 8]. Although this classification is well accepted in advanced GC, those of intramucosal neoplasia (IMN), including low-grade dysplasia (LGD), high-grade dysplasia (HGD), and intramucosal cancer (IMC), have not been fully elucidated.
In the present study, we stratified genetic differences using PCR-based microsatellite analysis [combination of allelic imbalance (AI) and MSI] to identify molecular patterns in IMNs. Here we present the specific molecular profiles of 158 IMNs treated with endoscopic submucosal dissection (ESD) and describe the value of this molecular approach for evaluation of early GC.
Materials and methods
Patients
One hundred fifty-eight patients with intestinal-type IMN who underwent endoscopic submucosal dissection and pathology examination at Iwate Medical University Hospital between 2010 and September 2014 were included. We evaluated the clinicopathological parameters by reviewing medical records and pathology records. Tumor histological classification was determined according to the modified WHO classification [13, 14]. Tumor staging was based on the classification of the Japanese Gastric Cancer Association [15]. None of the patients had fulfilled the revised Bethesda criteria for Lynch syndrome. The grade of the dysplasia was evaluated according to the modified WHO 2010 criteria [13]. LGD was characterized by a uniform monolayer of columnar cells with basal nuclei showing minimal atypia. In HGD, nuclear atypia was more frequent, with nuclear pleomorphism, nuclear enlargement, and pseudostratification without stromal invasion. In IMC there was marked cytological atypia and complex architecture, with cribriform groups, irregular branching, glandular anastomosis, and budding of neoplastic cells into the lumen, which were considered representative of stromal invasion. Tumors exhibiting a mixed pattern of IMC and HGD within the same tumor were termed “mixed tumors.” If papillary structures were found within a tumor, we increased the grade of the tumor because this structural atypia is found frequently in gastric adenocarcinomas. This study was approved by the Institutional Review Board of Iwate Medical University (“Clinicopathological and molecular findings of early gastric cancers based on tumor grade”; approval number H24-182).
For molecular investigations, tumor tissue was isolated from the resected stomach tissue by means of biopsy forceps within 30 min of resection. The most distant normal gastric mucosa from the neoplasm was removed from the submucosa by means of scissors as a control for molecular analysis. Tissue for clinicopathology analysis was obtained from a region of the resected stomach adjacent to the region used for molecular analysis. All other samples contained cancer tissue. Only tumor samples where the neoplastic cells accounted for at least 50 % of the tissue cell population were selected.
Immunohistochemistry
Immediately after excision, specimens were fixed in 20 % neutral buffered formalin, embedded in paraffin wax, cut into 4-μm-thick paraffin sections, and stained with hematoxylin and eosin for routine light microscopy [16]. For immunohistochemical staining, additional serial 3-μm-thick sections were cut from paraffin-embedded tissues and placed on poly(l-lysine)-coated glass slides. Briefly, sections were deparaffinized in xylene and dehydrated. For the determination of mucin and related protein expression, immunostaining was performed for mucin 2 (Ccp58; Novocastra Laboratories, Newcastle upon Tyne, UK), CD10 (56C6; Novocastra Laboratories), mucin 5AC (CLH2; Novocastra Laboratories), and mucin 6 (CLH5; Novocastra Laboratories). In addition, immunostaining was performed for p53 (clone DO7; DAKO, Carpinteria, CA, USA) and β-catenin.
Immunohistochemistry was performed with a DAKO EnVision+ system, with use of dextran polymers conjugated with horseradish peroxidase (DAKO), as previously described. The specimens were heated in citrate buffer (pH 6.0) three times for 5 min each in a microwave oven (H2500 microwave processor; Bio-Rad, Hercules, CA, USA) at 750 W before incubation with antibodies, as previously described [17]. Hematoxylin was used as the counterstain. Tumors were considered positive when more than 30 % of cells were immunopositive or negative when less than 30 % of cells were immunopositive.
Evaluation of the mucin phenotype
Early GCs were classified into four groups (gastric, intestinal, mixed, and unclassified) according to their immunostaining pattern. Determination of the mucin phenotype was performed as previously described [17]. If more than 10 % of the tumor cells were immunopositive, the tumor was regarded as positive for that antibody; otherwise the tumor was classified as negative.
PCR analysis
PCR was performed with a thermal cycler (GeneAmp PCR system 9600; PerkinElmer, Santa Clara, CA, USA) with 50–100 ng of genomic DNA as the template, each primer at a concentration of 25 pM , 0.2 mM deoxynucleotide triphosphate, 1× reaction buffer containing 1.5 mM MgCl2, and 1.5 U Taq polymerase (Boehringer Mannheim, Germany) in a final reaction volume of 25 μL. Samples were processed for 25–30 cycles, with each cycle consisting of 30 s at 94 °C, 1 min at 55–58 °C, and 2 min at 72 °C, followed by a final extension for 10 min at 72 °C. For quantitative detection of the AI at each locus, PCR–microsatellite analysis, including evaluation of AI and MSI, was performed as described previously [10].
Assessment of AI by PCR
We examined AIs at chromosomal loci 3p, 4p, 5q, 9p, 13q, 17p, 18p, and 22q in paired tumor and normal DNA samples obtained from 158 patients with early GC using 23 highly pleomorphic microsatellite markers (D1S228, D1S548, D1S507, D3S2402, D3S1234, D4S2639, D4S1601, D5S107, D5S346, D5S299, D5S82, D8S201, D8S513; D8S532, D9S171, D9S1118, D13S162, TP53, D18S487, D18S34, D22S274, D22S1140, and D22S1168). These microsatellite markers have been reported frequently in GCs. In addition, a variable number of tandem repeat polymorphisms at the DCC locus were tested. Microsatellite sequences were obtained from specific primers reported in the Genome Database (http://gdbwww.gdb.org/gdb/).
The peaks produced by the normal DNA sample were used to determine whether the cancerous sample was homozygous (one peak) or heterozygous (two peaks). The allelic ratio was calculated as described by Habano et al. [18]. A tumor was considered to have allelic loss if the allele peak ratio was less than or equal to 0.70, representing an allelic signal reduction of at least 30 %. Tumors exhibiting MSI at a given locus were not evaluated for AI. The data were collected automatically and analyzed with GeneScan (Applied Biosystems, Carlsbad, CA, USA) to determine the allele score and to assess the possibility of AI.
Assessment of MSI
Six different loci were considered for MSI assessment, including all those recommended by the Bethesda panel for CRC (i.e., BAT25, BAT26, D5S346, D2S123, and D17S250) [19]. Tumors were considered to be MSI positive when a novel, abnormally sized band occurred in the tumor sample as compared with the corresponding normal DNA sample. MSI-positive CRCs were used as controls and were divided into three groups. MSI status was determined by five markers described by the National Cancer Institute: BAT25, BAT26, D2S123, D5S346, and D17S250. MSI-high samples were defined as those having two or more unstable markers, MSI-low samples were defined as those having one unstable marker, and MSS samples were defined as those having no instability. Samples that were previously categorized as MSI low and MSS were considered MSI-negative tumors in this study.
Pyrosequencing for evaluation of methylation
The DNA methylation status of each gene promoter region was established by PCR analysis of bisulfite-modified genomic DNA (EpiTect bisulfite kit; Qiagen) by means of pyrosequencing for quantitative methylation analysis (PyroMark Q24; Qiagen) [20]. The primers were designed with the PyroMark assay designing software package (Qiagen), with three to four CpG sites included for analysis of promoter methylation.
DNA methylation was quantified at six specific promoters originally described by Yagi et al. [21, 22]. Briefly, after methylation analysis of a panel of three markers (RUNX3, MINT31, and LOX), tumors with a high-methylation epigenotype (HME) were defined as those with at least two methylated markers. The remaining tumors were examined with use of three markers (NEUROG1, ELMO1, and THBD). Tumors with an intermediate-methylation epigenotype (IME) were defined as those with at least two methylated markers, and tumors not classified as having an HME or an IME were designated as having a low-methylation epigenotype (LME).
The cutoff value for the mutation assay was 15 % mutant alleles, whereas that for the methylation assay was 30 % of tumor cells, as previously reported.
Statistical analysis
Hierarchical analysis was performed for clustering of the samples according to MSI and AIs to achieve maximal homogeneity for each group and the greatest difference between the groups with use of open-access clustering software (Cluster 3.0; http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). The clustering algorithm was set to centroid linkage clustering, which is the standard hierarchical clustering method used in biological studies.
Data were analyzed with Stat Mate-III (Atom, Tokyo, Japan). Data obtained for histological features, immunohistochemical findings, AI of chromosomal loci, MSI status, and methylation status based on each subgroup were analyzed by chi-square tests. If statistical differences between four groups were found, statistical analyses between two groups were further performed by chi-square tests. Differences in age distributions among patients in the four groups were evaluated by the Kruskal–Wallis H test. Differences with p values of less than 0.05 were considered significant.
Results
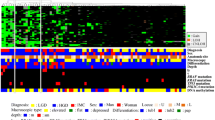
We performed hierarchical clustering based on the AI and MSI status to evaluate differences in genetic alterations in patients with IMN. Four distinct, albeit somewhat overlapping, clusters emerged, as shown in Fig. 1. The vertical line shows genetic alterations (AI or MSI), and the horizontal lines denote “relatedness” between samples.

Hierarchical analysis of intramucosal neoplasia using PCR-based microsatellite analysis. The intramucosal neoplasias examined were subclassified into four subgroups. AI allelic imbalance, IHCs immunohistochemical staining, MSI, microsatellite instability
Clinicopathology findings are depicted in Table 1. There were no significant differences in terms of age, sex, tumor location, and macroscopic type between the four subgroups. However, significant differences in papillary adenocarcinoma type between subgroup 1 and subgroups 2, 3, and 4 were found (p < 0.01). The following histological features are suggestive of EBV-associated GC: (1) a lace pattern of the cancer cells within the mucosa and (2) a lymphoepithelioma-like lesion within the mucosa or submucosa [23]. These two findings were not observed in the IMNs examined. Whereas the frequency of LGD was higher in subgroup 4 than in subgroups 1 and 3 (p < 0.01 for both), that of HGD was higher in subgroup 1 than in subgroups 2 and 4 (p < 0.05 for both). In addition, there were significant differences in the frequencies of IMCs between subgroups 2 and 4 and between subgroups 3 and 4 (p < 0.05 for both).
Tumors with MSI-high status were subclassified into subgroup 1 (20 of 21 subgroup 1 tumors), subgroup 2 (5 of 19 subgroup 2 tumors), and subgroup 4 (2 of 85 subgroup 4 tumors), as shown in Fig. 1, whereas tumors with MSI-low status were subdivided into subgroup 2 (three MSI-low tumors) and subgroup 4 (one MSI-low tumor). Tumors exhibiting microsatellite stability were mainly assigned to subgroup 3 (33 MSS tumors) and subgroup 4 (82 MSS tumors). In addition, MC was more frequently found in subgroup 3 than in subgroup 4 (p < 0.05). There were significant differences in MSI-high status between subgroup 1 and subgroups 2, 3, and 4 (p < 0.01).
Tumors with multiple AIs were allocated to subgroup 3, as shown in Fig. 1. Multiple AIs were found in subgroup 3 compared with subgroup 4, and there were significant differences in the frequencies of individual loci between subgroups 3 and 4, as listed in Table 2. The frequency of AI at 8p was significantly higher in subgroup 3 than in the other three groups (p < 0.01). In addition, 17p AI was more frequent in subgroups 1 and 3 than in subgroup 4 (Table 2, p < 0.05 and p < 0.001 respectively), and 22q AI was more frequently found in subgroup 2 than in subgroup 4 (p < 0.01). On the other hand, the frequencies of AIs at the chromosomal loci examined were lower in subgroup 4 than in subgroup 3. Subgroup 2 contained tumors with AIs and MSI, indicating an intermediate category between subgroups 1 and 3.
There were significant differences in the frequencies of mucin 5AC or mucin 6 expression in subgroup 1 compared with subgroups 3 and 4 (p < 0.01). On the other hand, CD10 expression was more frequent in subgroup 2, 3, or 4 than in subgroup 1 (p < 0.01). The frequency of nuclear β-catenin expression differed significantly between subgroups 1 and 3 (p < 0.05). MLH1 expression was significantly reduced in subgroup 1 compared with that in subgroups 2, 3, and 4 (p < 0.001 for all comparisons). In addition, MLH1 loss was more frequent in subgroup 2 than in subgroups 3 and 4 (p < 0.05). Overexpression of p53 was found in all subgroups (Table 3).
Fourteen (66.7 %) of the 21 tumors in subgroup 1 displayed an HME pattern, and no tumors in subgroup 1 showed an LME pattern. In contrast, 8 (24.2 %) of 33 subgroup 3 tumors and 17 of the 85 subgroup 4 tumors demonstrated an HME pattern. The differences in HME patterns between subgroup 1 and subgroup 3 or 4 were statistically significant (p < 0.01 for both; Fig. 2). In addition, the frequency of HME in subgroup 2 was significantly higher than that in subgroup 4 (p < 0.05). There were significant differences in the frequencies of IME between subgroup 2 and subgroups 3 and 4 (p < 0.05 for both). Furthermore, subgroup 3 showed a higher frequency of IME than subgroup 1 (p < 0.05). Finally, the frequency of LME in subgroup 4 was significantly higher than that in subgroup 1 (p < 0.05). Representative images of MSS and MSI IMNs are shown in Figs. 3 and 4.

Frequencies of methylation status in subgroups stratified by unsupervised hierarchical analysis. HME high-methylation epigenotype, IME intermediate-methylation epigenotype, LME low-methylation epigenotype

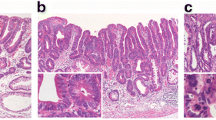
Analysis of histological features, mucin phenotype, and genetic alterations in microsatellite-stable cancers. a Histological features of differentiated-type adenocarcinoma. b No expression of mucin 5AC (MUC5AC). c Tumor cells focally expressing mucin 6 (MUC6). d No expression of mucin 2 (MUC2). e Tumor cells strongly expressing CD10. g Tumor cells overexpressing p53. h Intranuclear expression of β-catenin. i LOX, MINT31, and RUNNX3 were methylated, indicating the high-methylation phenotype. j Electrophoregram demonstrating allelic imbalances at chromosomal loci 5q, 17p, and 18q. k BAT25 and BAT26 showed microsatellite stabilty status. MSI microsatellite instability, N normal, T tumor

Analysis of histological features, mucin phenotype, and genetic alterations in microsatellite-instable cancers. a Histological features of papillary adenocarcinoma. b Tumor cells strongly expressing mucin 5AC (MUC5AC). c Tumor cells focally expressing mucin 6 (MUC6). d, e No expression of mucin 2 (MUC2) and CD10. f No tumor cells expressed (MLH1). g No overexpression of p53. h No intranuclear expression of β-catenin. i LOX, MINT31, and RUNX3 were methylated, indicating the a high-methylation phenotype. j Electrophoregram demonstrating allelic imbalances at 5q. k BAT25 and BAT26 showed microsatellite instability (MSI) status. N normal, T tumor
Discussion
GC can be divided into adenocarcinomas of diffuse and intestinal types according to the Lauren classification [24]. Intestinal-type GCs are often observed in the context of Helicobacter pylori-associated chronic gastritis, atrophy, and intestinal metaplasia, which may be precursors of the dysplastic changes that evolve into this type of carcinoma (or paracancerous lesion) [16]. Diffuse-type GCs are also associated with H. pylori infection, but arise independently of intestinal metaplasia [24]. These types of GC are usually less well differentiated, characterized by sheets of cells without gland formation, with the occasional presence of signet ring cells, and are associated with a poor prognosis compared with intestinal-type GCs [24]. Therefore, intestinal-type adenocarcinoma is thought to be distinct from diffuse-type adenocarcinoma with respect to gastric carcinogenesis [16, 24]. In the present study, intestinal-type IMNs, including differentiated-type GC, were the main target lesions used to identify molecular characteristics of early gastric carcinogenesis.
Hierarchical cluster analysis has been used to stratify genetic differences in many tumors to identify their genetic characteristics. This method has been used for identification of genome-wide alterations, which is usually achieved by array-based analysis (e.g., array comparative genomic hybridization or SNP array analysis), an expensive and laborious technique [8]. However, in the present study, we stratified IMNs by PCR-based microsatellite analysis using a limited number of markers at cancer-related chromosomal loci that are closely associated with gastric carcinogenesis to examine genetic differences in IMNs. This method was simple, inexpensive, and useful for examination of genetic differences in the samples and only a small amount of tumor tissue was required. In addition, this method could be used to objectively analyze genetic differences in the tumors. We showed that genetic differences stratified by hierarchical cluster analysis using a combination of AI and MSI status may help identify patients with IMN who are likely to experience malignant transformation.
Genetic distinctions between subtypes resulting from different etiological pathways may translate to differences in tumor behavior [3, 4, 7, 8]. As tumor markers that can reflect different genetic pathways, AI, MSI, CIMP, and p53 status have been studied extensively in advanced GCs [3, 4, 25, 26]. However, these characteristics have not been clearly defined in early GC. Therefore, a better understanding of the underlying molecular alteration in IMNs is required. Among these molecular alterations, AIs and MSI status represent direct tumor chromosomal damage that plays an important role in tumor progression [3, 7, 20]. This approach will provide essential and objective information for predicting tumor invasion or progression. Our data showed that tumors stratified by genetic alterations based on AIs and MSI status could be classified into distinct subgroups; this classification indicated that IMNs are heterogeneous tumors that develop via acquisition of different molecular profiles involving a combination of genetic and epigenetic changes. In addition, we found that early gastric tumorigenesis was closely associated with different biomarkers.
In the present study, tumors in subgroup 1 were characterized by MSI-high status. Previous studies have shown that most MSI-high tumors are of the intestinal histotype, located in the distal portion of the stomach, and occur more frequently in older women [27]. In particular, MSI-high tumors show distinct histological features, such as mucinous, serrated, and medullary patterns, in advanced cancers [27]. In the present study, tumors with a papillary structure were frequently found in subgroup 1 IMNs, indicting MSI-high status. Papillary structure may help predict early detection of IMNs with MSI-high status [28].
CIN is considered a representative genetic pathway in gastric carcinogenesis [6–8]. The most prominent finding in the CIN pathway is that chromosomal alterations are frequently found in the tumors [6–8]. In addition, CIN has been shown to occur primarily in advanced GC [6, 7]. However, the role of CIN in IMN is not fully understood. In the present study, approximately 20 % of the IMNs examined were classified as having CIN, which was used to define subgroup 3. This finding supports the hypothesis that tumors classified into subgroup 3 had a higher risk of progression to invasive cancer than tumors without CIN, which were characterized by low frequency of AI and MSI-negative/MSI-low status, consistent with the observation that CIN is responsible for malignant transformation of IMN to advanced GC.
In the present study, we found that approximately 50–60 % of IMNs classified into subgroup 4 were characterized by low AI and negative MSI, suggesting that there was also a distinct molecular profile for tumors having neither CIN status nor MSI status in early-stage GC. Furthermore, the present study indicated that DNA methylation was not a genome-level change involved in the development of early-stage GC. Although we cannot rule out the possibility of mutations or other chromosomal alterations in tumor cells, our data suggested that epigenetic changes may be essential for development of IMN.
Genome-wide DNA methylation status is known to play a major role in gastric carcinogenesis [9]. Genome-wide DNA methylation status (CIMP) has been assessed by the one-panel method developed by Toyota et al. [9]. According to their study, genome-wide DNA methylation status is subclassified into two subgroups: CIMP high and CIMP low. Kaneda and Yagi [22] developed a two-panel method and subclassified tumors into three groups: HME, IME, and LME. Yagi et al. [21, 22] showed that the two-panel method reflects the status of genome-wide DNA methylation in CRC cells. Their findings demonstrated that although HME is strongly correlated with MSI-high status in CRC, IME is characterized by KRAS mutations in CRCs [21, 22]. In addition, they emphasized that IME is correlated with poor prognosis in patients with CRC [21, 22]. Although it remains unclear whether the two-panel method can reflect the genome-wide DNA methylation status of the gastric tumor cells, we attempted to identify the role of three subgroups of DNA methylation (HME, IME, and LME) in gastric IMNs. In the present study, there were significant differences in the frequencies of HME between subgroup 1, which was characterized by MSI-high status, and the other groups. This finding is consistent with those of previous studies [29]. In addition, we showed that there was a close association between the IME and subgroup 3, in which multiple AIs were accumulated. We also demonstrated that the IME played an important role in subgroup 4 tumors during early gastric tumorigenesis. This is interesting because the role of IME has been identified in specific types of GCs and IME is a common alteration occurring during the precancerous stage of gastric tumors. Thus, we suggest that the two-panel method may accurately reflect the genome-wide DNA methylation status of gastric IMN cells.
Differences in diagnoses provided by Japanese and Western pathologists are known to occur because of discrepancies in the histological criteria assigned to the intramucosal neoplastic lesion [30]. This serious diagnostic difference between Japanese and Western pathologists has not been clearly resolved. Although previous studies have indicated that multiple genomic changes reflect tumor aggressiveness or invasion [7], the significance of subtype classification based on combinations of these two markers (AI and MSI) with respect to the aggressiveness or invasion of gastric tumors has not been well studied. In the present study, multiple AIs and MSI were found in not only IMCs or mixed tumors but also some HGDs. This finding supports the observation that at least half of HGDs have malignant potential and should be treated as cancerous.
Previous studies have shown that mucin expression in tumor cells plays a major role in gastric carcinogenesis [16, 17]. In the present study, whereas the MSI-high phenotype (subgroup 1) was categorized by the expression of either mucin 5AC or mucin 6, phenotypic expression in subgroup 3 or subgroup 4 was closely associated with CD10 expression. Our findings suggested that although the mucin phenotype (e.g., gastric, intestinal, mixed phenotypes) was not correlated with a specific subgroup in the present study (data not shown), differences in the biological behaviors of tumors with different individual mucin markers may result from genetic backgrounds during the incipient phase of gastric tumorigenesis.
Changes in p53 status (e.g., p53 overexpression or mutation) play a major role in gastric carcinogenesis [7, 8]. Alterations in p53 are known to be influenced by different factors, including tumor histological features, tumor grade, tumor stage, and mucin expression [16, 17]. Previous studies have shown that p53 overexpression is associated with accumulation of chromosomal alterations, but is an infrequent alteration in MSI-high tumors [7]. In the present study, however, we provided evidence to support that p53 overexpression is a fundamental characteristic of gastric carcinogenesis, irrespective of the accumulation of AIs.
Nuclear accumulation of β-catenin often occurs in CRC and other cancers [29]. Subsequent changes in the nuclear expression of β-catenin are thought to contribute to tumor initiation in colorectal tumorigenesis [31]. Activation of the Wnt/β-catenin signaling pathway is found in about 30–50 % of GC tissues and in many GC cell lines [32]. The level of β-catenin in the nucleus is an indicator of an active Wnt signal pathway and is expected to be a useful biomarker associated with disease progression in GC and CRC [31, 32]. However, the role of β-catenin in gastric carcinogenesis is not fully understood. In the present study, our data suggested that nuclear accumulation of β-catenin was associated with the development of subgroup 3 tumors, which were thought to exhibit microsatellite stability (CIN).
The Cancer Genome Atlas (TCGA) is a comprehensive study that has attracted the attention of many researchers in the field of gastric carcinogenesis [8]. According to current theory, GC can be classified into four subgroups (i.e., MSI, EBV-positive, CIN, and GS GC), as mentioned already [8]. Thus, it may be useful to describe how the present subgroups can be classified within the four subgroups described by TCGA. In the present study, although subgroup 1 was assigned to the MSI-high phenotype, subgroup 3 corresponded to the CIN phenotype. Although subgroup 4 was closely associated with low frequencies of AI and MSI and exhibited a pattern of AI and MSI similar to that observed in the GS phenotype, this subgroup could not be simply applied to the GS type because GS tumors are histologically characterized as diffuse-type tumors with high frequencies of CDH1 (which encodes E-cadherin) and RHOA mutations [8]. In addition, diffuse-type GC was not observed in the IMNs examined in this study. Notably, in the present study, characteristic histological features, including a lace pattern of the cancer cells within the mucosa and a lymphoepithelioma-like lesion with the mucosa or submucosa [23, 33], were not found in the IMNs examined. Although we could not examine the presence/absence of EBV in the cancer cells in the present study, we speculate that EBV-associated GC was not involved in the IMNs we examined. We suggest that the present subgroups conformed to the subtypes described by TCGA.
In conclusion, we examined the genetic differences in IMNs by stratifying the genetic alterations obtained from analysis of AIs and MSI. These data provide important insights into the molecular mechanisms responsible for the development of IMNs. Furthermore, our data support the notion that PCR-based microsatellite analysis using a limited number of chromosomal loci can identify distinct subgroups in IMNs.
References
Valente P, Garrido M, Gullo I, Baldaia H, Marques M, Baldaque-Silva F, et al. Epithelial dysplasia of the stomach with gastric immunophenotype shows features of biological aggressiveness. Gastric Cancer. 2015;8:720–8.
Shi J, Qu YP, Hou P. Pathogenetic mechanisms in gastric cancer. World J Gastroenterol. 2014;20:13804–19.
Ottini L, Falchetti M, Lupi R, Rizzolo P, Agnese V, Colucci G, et al. Patterns of genomic instability in gastric cancer: clinical implications and perspectives. Ann Oncol. 2006;17(Suppl 7):vii97–102.
Hiyama T, Tanaka S, Yoshihara M, Sasao S, Kose K, Shima H, et al. Chromosomal and microsatellite instability in sporadic gastric cancer. J Gastroenterol Hepatol. 2004;19:756–60.
Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9.
Liang L, Fang JY, Xu J. Gastric cancer and gene copy number variation: emerging cancer drivers for targeted therapy. Oncogene. 2015. doi:10.1038/onc.2015.209.
Sugai T, Habano W, Endoh M, Konishi Y, Akasaka R, Toyota M, et al. Molecular analysis in gastric differentiated-type intramucosal and submucosal cancers. Int J Cancer. 2010;127:2500–9.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6.
Sugai T, Habano W, Nakamura S, Sato H, Uesugi N, Takahashi H, et al. Genetic alterations in DNA diploid, aneuploid and multiploid colorectal carcinomas identified by the crypt isolation technique. Int J Cancer. 2000;88:614–9.
Jiao Y-F, Sugai T, Habano W, Suzuki M, Takagane A, Nakamura S. Analysis of microsatellite alterations in gastric carcinoma by applying the crypt isolation technique. J Pathol. 2004;204:200–7.
Sugai T, Habano W, Jiao Y-F, Suzuki M, Takagane A, Nakamura S. Analysis of genetic alterations associated with DNA diploidy, aneuploidy and multiploidy in gastric cancers. Oncology. 2005;68:548–57.
Lauwers GY, Carneiro F, Graham DY, Bosman FT, Carneiro F, Hruban RH, et al. Gastric carcinoma. WHO classification of tumours of the digestive system. Lyon: International Agency for Research on Cancer; 2010. p. 48–58.
Sakurai U, Lauwers GY, Vieth M, Sawabe M, Arai T, Yoshida T, et al. Gastric high-grade dysplasia can be associated with submucosal invasion: evaluation of its prevalence in a series of 121 endoscopically resected specimens. Am J Surg Pathol. 2014;38:1545–50.
Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer. 2011;14:101–12.
Sugai T, Tsukahara M, Endoh M, Shioi Y, Takebe N, Mue Y, et al. Analysis of cell cycle-related proteins in gastric intramucosal differentiated-type cancers based on mucin phenotypes: a novel hypothesis of early gastric carcinogenesis based on mucin phenotype. BMC Gastroenterol. 2010;10:55.
Sugai T, Habano W, Uesugi N, Jiao Y-F, Nakamura S, Abe K, et al. Three independent genetic profiles based on mucin expression in early differentiated-type gastric cancers—a new concept of genetic carcinogenesis of early differentiated-type adenocarcinomas. Mod Pathol. 2004;17:1223–34.
Habano W, Sugai T, Nakamura S, Yoshida T. A novel method for gene analysis of colorectal carcinomas using a crypt isolation technique. Lab Invest. 1996;74:933–40.
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57.
Yamamoto E, Suzuki H, Yamano HO, Maruyama R, Nojima M, Kamimae S, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol. 2012;181:1847–61.
Yagi K, Takahashi H, Akagi K, Matsusaka K, Seto Y, Aburatani H, et al. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol. 2012;180:616–25.
Kaneda A, Yagi K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci. 2011;102:18–24.
Fukayama M. Epstein–Barr virus and gastric carcinoma. Pathol Int. 2010;60:337–50.
Berlth F, Bollschweiler E, Drebber U, Hoelscher AH, Moenig S. Pathohistological classification systems in gastric cancer: diagnostic relevance and prognostic value. World J Gastroenterol. 2014;20:5679–84.
Kim KJ, Lee TH, Cho NY, Yang HK, Kim WH, Kang GH. Differential clinicopathologic features in microsatellite-unstable gastric cancers with and without MLH1 methylation. Hum Pathol. 2013;44:1055–64.
Vauhkonen M, Vauhkonen H, Sajantila A, Sipponen P. Differences in genomic instability between intestinal- and diffuse-type gastric cancer. Gastric Cancer. 2005;8:238–44.
Arai T, Sakurai U, Sawabe M, Honma N, Aida J, Ushio Y, et al. Frequent microsatellite instability in papillary and solid-type, poorly differentiated adenocarcinomas of the stomach. Gastric Cancer. 2013;16:505–12.
Sugimoto R, Sugai T, Habano W, Endoh M, Eizuka M, Yamamoto E, et al. Clinicopathological and molecular alterations in early gastric cancers with the microsatellite instability-high phenotype. Int J Cancer. 2016;138:1689–97.
An C, Choi IS, Yao JC, Worah S, Xie K, Mansfield PF, et al. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res. 2005;11:656–63.
Schlemper RJ, Kato Y, Stolte M. Review of histological classifications of gastrointestinal epithelial neoplasia: differences in diagnosis of early carcinomas between Japanese and Western pathologists. J Gastroenterol. 2001;36:445–56.
Kennell J, Cadigan KM. APC and β-catenin degradation. Adv Exp Med Biol. 2009;656:1–12.
Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009;5:e1000676.
Song HJ, Srivastava A, Lee J, Kim YS, Kim KM, Ki Kang W, et al. Host inflammatory response predicts survival of patients with Epstein–Barr virus-associated gastric carcinoma. Gastroenterology. 2010;139:84–92.
Acknowledgments
We gratefully acknowledge the technical assistance of E. Sugawara and T. Kasai. We also thank members of the Department of Molecular Diagnostic Pathology, Iwate Medical University, for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Statement of author contributions
T. Sugai contributed to the preparation of the manuscript, including all aspects of data collection and analysis. R. Sugimoto constructed the figures and tables and performed statistical analysis. W. Habana, T. Matsumoto, and H. Suzuki provided input during the preparation of the manuscript. E. Yamamoto, M. Eizuka, and N. Arakawa provided support for experiments involving molecular technologies. M. Endoh, K. Kawasaki, and S. Yanai assisted with clinical data and experiments.
Human rights statement and informed consent
All procedures followed were in accordance with the ethical standards of Iwate Medical University and with the Declaration of Helsinki. Substitute for informed consent (approval of the Institutional Review Board of Iwate Medical University) was obtained from all patients for their being included in the study. This study was approved by the Institutional Review Board of Iwate Medical University (“Clinicopathological and molecular findings of early gastric cancers based on tumor grade”; approval no. H24-182).
Rights and permissions
About this article

Cite this article
Sugai, T., Sugimoto, R., Habano, W. et al. Genetic differences stratified by PCR-based microsatellite analysis in gastric intramucosal neoplasia. Gastric Cancer 20, 286–296 (2017). https://doi.org/10.1007/s10120-016-0616-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-016-0616-2