
Abstract
Coronaviruses are a group of envelop viruses which lead to diseases in birds and mammals as well as human. Seven coronaviruses have been discovered in humans that can cause mild to lethal respiratory tract infections. HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1 are the low-risk members of this family and the reason for some common colds. Besides, SARS-CoV, MERS-CoV, and newly identified SARS-CoV-2, which is also known as 2019-nCoV, are the more dangerous viruses. Due to the rapid spread of this novel coronavirus and its related disease, COVID-19, a reliable, simple, fast, and low-cost detection method is necessary for patient diagnosis and tracking worldwide. Human coronaviruses detection methods were classified and presented in this article. The laboratory detection techniques include RT-PCR, RT-LAMP, electrochemical and optical biosensors for RNA detection, and whole virus or viral proteins detection assays.
Similar content being viewed by others

Introduction
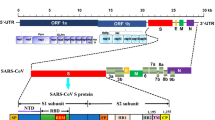
Coronaviruses are members of the Coronaviridae family, which belongs to the Nidovirales order. These viruses are enveloped, non-segmented, positive-sense, and single-stranded RNA viruses that cause mild or severe diseases in some birds and mammals, including humans. Their genome size is about 30 kilobases (kb) which consist of non-structural open reading frames (ORFs) near the 5′-end and at least four structural proteins near the 3′-end, including membrane (M), envelope (E), spike (S), and nucleocapsid (N) proteins. The coronavirus name derives from their solar corona appearance, which is formed due to the club-shaped spikes that project from the surface of the virion [1,2,3,4].
The first study on coronaviruses was reported in 1931, while the first human coronaviruses were identified in the 1960s. HCoV-229E and HCoV-OC43 were discovered as responsible viruses for some cases of cold and respiratory tract infection. According to their genome structure, HCoV-229E is classified in the alpha-coronaviruses (or group 1) subgroup while HCoV-OC43 belongs to beta-coronaviruses (or group 2) subgroup [5,6,7,8,9].
Severe acute respiratory syndrome (SARS) was reported for the first time in November 2002 in China, and SARS-associated coronavirus (SARS-CoV) was identified in March 2003 as the third human coronavirus. This dangerous virus was characterized and classified in the beta-coronaviruses subgroup. SARS outbreak involved 29 countries, infected over 8000 people, and caused 774 deaths (the fatality rate was about 9%). There has not been any new SARS case reported since 2004 [2, 10, 11].
After the SARS epidemic, several research investigations have been done to find the new human coronaviruses. HCoV-NL63 was reported in 2004 in the Netherlands as a member of alpha-coronaviruses. Afterward, the next human beta-coronavirus was identified in 2005, which was named HCoV-HKU1. In a few years after the discovery of HCoV-NL63 and HCoV-HKU1, numerous reports were published about the presence of these coronaviruses in patients with both upper and lower respiratory tract infections worldwide [12,13,14].
The sixth human coronavirus was emerged in the Middle East in 2012 and named Middle East respiratory syndrome-CoV (MERS-CoV). MERS-CoV is another member of the beta-coronaviruses subgroup and could infect humans and dromedary camels. So, dromedary camels are known as the possible zoonotic source for MERS-CoV. Furthermore, MERS was a severe lower respiratory tract infection with a high fatality rate similar to SARS. A total of 2519 MERS confirmed cases were reported until the end of January 2020, which has led to 866 deaths, with a case-fatality rate of 34.3% [2, 15, 16].
The last human coronavirus appeared in December 2019 in Wuhan, China, which was named 2019 novel coronavirus (2019-nCoV) by the World Health Organization (WHO) and then renamed to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), due to its similarity to SARS-CoV. Also, WHO named the related disease as coronavirus disease 2019 (COVID-19). The ongoing pandemic is growing fast, and between 100,000 and 300,000 new cases per day were reported over the past month. Furthermore, about sixteen million laboratory-confirmed cases around the world and more than 600,000 deaths have been reported until 25th July 2020 [17,18,19,20,21].
Due to the rapid spread of COVID-19, a reliable detection method is needed for patient diagnosis and tracking worldwide, especially in the early stages of the disease, to slow and try to control the pandemic. WHO has recommended nucleic acid amplification tests (NAAT), such as reverse transcription polymerase chain reaction (RT-PCR), for this purpose [22].
Nevertheless, the RT-PCR method is a relatively expensive and time-consuming technique and needs an expert technician, as well as specialized equipment compared to other diagnostic methods. It has also shown some false-negative results (even more than 30%) in COVID-19 diagnosis compare to chest CT imaging technique results. Furthermore, except chest CT-imaging and host antibody detection (serological assays) that have been used for diagnosis purposes, there are other methods for SARS-CoV-2 direct detection, such as RT-LAMP, viral RNA biosensing, and viral protein biosensors [23,24,25,26,27,28].
In this article, human coronaviruses laboratory detection methods allowing direct virological diagnosis are separated in three classes: RNA amplification-based detection methods (including RT-PCR, real-time RT-PCR, and isothermal amplification-based methods), viral RNA biosensors (including electrochemical and optical biosensors), and whole virus or viral proteins detection assays.
It should be noted that the technical point of view was selected in the article to introduce the potentially applicable techniques for fighting against COVID-19 pandemic. As all the human coronaviruses have a relatively similar genomes, the previously used techniques may be suitable for SARS-CoV-2. So, the methods were classified based on the detection principles. Nevertheless, the genetic divergence between the seven human coronaviruses, including the viruses classified in the same subgroup, leads to the use of specific systems for each virus.
Figure 1 represents the timeline of the introduction of these techniques regarding each of the seven human coronaviruses. Moreover, several commercial real-time RT-PCR-based and antigen-specific COVID-19 diagnostic kits are discussed.

The timeline of the introduction of the diagnostic methods regarding each of the seven human coronaviruses
RNA amplification-based detection methods
Most of the viral RNA detection methods work based on nucleic acids amplification. As the amplification methods are usually used for DNA duplication, the complementary DNA (cDNA) of the target must be formed. Therefore, reverse transcription of the extracted target RNA to form the cDNA is the first step in the RNA amplification-based detection methods. Then, the target cDNA is multiplied in the amplification step. In the initially presented methods, the target cDNA was detected after the end of the amplification step. Although, the real-time detection methods were then developed, and hence, amplification and detection steps were merged, some highly sensitive methods were also proposed to directly detect the target RNA without amplification [29,30,31].
Different RNA detection formats are shown in Fig. 2. The total detection time of direct RNA detection methods is usually lower than that of the amplification-based techniques.

Different RNA detection formats
Table 1 summarizes the RNA amplification-based assays developed for the detection of coronaviruses, and the related studies are presented in the next three subsections.
Reverse transcription polymerase chain reaction (RT-PCR)
The polymerase chain reaction (PCR) technique is similar to a molecular photocopier that can synthesize a large number of a specific DNA sequence. The schematic illustration of PCR process is shown in Fig. 3. In this technique, a mixture of sample DNA, primers, nucleotides, and DNA polymerase brings in a three-step temperature cycle, which consists of denaturation, primer hybridization, and extension. After increasing the number of DNA molecules, it could be detected by various methods like agarose gel electrophoresis [31, 81, 82].

Schematic illustration of PCR process
Reverse transcription PCR (RT-PCR) technique was developed for specific RNA detection. In this method, the target RNA is converted to cDNA by the reverse transcriptase enzyme. Then, the obtained cDNA is amplified by PCR. In the traditional RT-PCR detection methods, the detection step is performed separately after the amplification step.
Thereafter, the amplification and detection steps have been combined together to decrease the total detection time and increase the sensitivity and accuracy of the method. This technique is known as real-time RT-PCR (rRT-PCR) or quantitative RT-PCR (qRT-PCR) and is represented in the next subsection [31, 83, 84].
Vabret et al. published the first research on coronaviruses detection in 2001, which was based on the RT-PCR method. They developed an RT-PCR assay for HCoV-229E and HCoV-OC43 and used agarose gel electrophoresis and DNA enzyme immunoassay for the detection of PCR products. They tested their method on 348 sputum and nasal aspirate samples, which could detect 0.05 median tissue culture infectious dose (TCID50)/ml of HCoV-229E and 0.01 TCID50/ml of HCoV-OC43 [32]. Vallet et al. carried out another study on coronaviruses detection in 2004. They investigated 2028 clinical nasal specimens for HCoV-229E by the RT-PCR method. The RT-PCR products (amplicon) were detected by a DNA enzyme immunoassay, which used a horseradish peroxidase (HRP)-labeled anti-double-stranded DNA to detect DNA hybridization. Their large-scale experiments showed 0.5 TCID50/ml detection limit, and the total detection time of each test was about 10 h [33].
Moës et al. reported an RT-PCR detection assay for HCoV-NL63, HCoV-OC43, and HCoV-229E, which were used for testing 309 nasal aspirates in 2004. After the termination of the RT-PCR process, the amplicon was sequenced by commercial DNA sequencer and compared with standard sequences. Also, the limit of detection (LOD) of their method was 5000 copies/μl and 50 copies/μl for HCoV-NL63 and HCoV-OC43, respectively [34]. In 2007, Lam and teammates used a fast thermo-cycler and developed a multiplex RT-PCR for the detection of 21 respiratory viruses consisting of SARS-CoV, HCoV-OC43, and HCoV-229E. They tested 303 nasal aspirate samples, and the amplicon was detected by agarose gel electrophoresis. The total detection time was about 5 h and the LOD was 10 copies/reaction for all the three coronaviruses [35].
To decrease the detection time and simplify the operation, Thaitrong and coworkers designed an integrated capillary electrophoresis microsystem and combined the RT-PCR and electrophoresis separation steps. They applied the designed microsystem to detect different respiratory viruses including HCoV-OC43 in spiked RNA solutions. This system was able to measure the RNA concentration in the range of 25–10,000 copies/reaction within 2 h and the LOD was 10 copies/reaction [36].
In another research, Shi et al. by using RT-PCR, reported an optical detector based on surface plasmon resonance (SPR), which identified nine common respiratory viruses, including SARS-CoV. The amino-functionalized oligonucleotide probes specific to each virus were immobilized on the 12-mercaptododecanoic-modified gold chips. Amplified extracted viral RNA sequences from throat swab samples were detected via the hybridization reaction occurring on the chips’ surface. The assay took about 2 h and a detection limit of 2 nM for SARS-specific viral RNA was observed. The downside to this work was the required cumbersome step to amplify the extracted RNA, which was due to the assay’s low sensitivity [37].
Real-time RT-PCR (rRT-PCR)
As previously mentioned, the incorporation of amplification and detection steps is a suitable way to decrease the detection time. After the development of fluorescent DNA labeling technique, real-time RT-PCR emerged. Therefore, by increasing the number of cDNA molecules in the mixture, the emitted fluorescent light increased. SYBR Green dye is a non-specific fluorescent DNA label that is widely used in rRT-PCR detection. SYBR Green binds to any double-strand DNA and light is emitted after binding. In clinical diagnosis, there have been some false-positive results due to the binding of SYBR Green dye to primer-dimers or other DNA double strands in the mixture [31, 82]. To resolve this problem, specific fluorescent probes, also known as reporters were developed, which only worked in the presence of a specific DNA sequence. The most common specific probe is the TaqMan probe, and methods based on this probe have been recommended as the standard diagnostic protocol for SARS-CoV-2 detection by WHO. In this method, fluorescence resonance energy transfer (FRET) is exploited and a fluorescent dye molecule and a quencher attached to 5′ end and 3′ end of the oligonucleotides. The quencher and fluorescent components are closed in the same molecule at first, and light emission is prevented. After cDNA amplification, the probe is hybridized to the specific sequence of single-stranded cDNA. In the extension step of PCR, the hybridized probe is split. Thus, the fluorescent and quencher components are separated which results in the increase of fluorescent intensity [85,86,87].
The rRT-PCR technique is the most common method in coronaviruses detection, due to its high sensitivity and accuracy, as well as relatively low detection time. Escutenaire et al. used SYBR Green probe to develop an rRT-PCR assay to detect SARS-CoV, HCoV-OC43, HCoV-229E, HCoV-NL63, and even 32 different animal coronaviruses. The detection time was almost two and half hours, and they could detect 10 copies/reaction of SARS-CoV [40].
Poon and colleagues reported an rRT-PCR assay in 2004 for detection of SARS-CoV by using TaqMan probe in 86 nasopharyngeal aspirate specimens. The assay concentration range was 10–106 copies/reaction, and the sensitivity and specificity of their tests were 86.2% and 100%, respectively [38]. Vijgen et al. performed the other TaqMan-based rRT-PCR assay in 2005 for HCoV-OC43 and HCoV-229E. They compared the TaqMan-based rRT-PCR assay with conventional RT-PCR by electrophoresis detection for 100 respiratory specimens including nasopharyngeal aspirates, sputum samples, bronchial aspirates, bronchoalveolar lavage specimens, and pharyngeal swabs. The diagnosis rate of the real-time assay was 50% and 700% higher than that of the conventional RT-PCR for HCoV-OC43 and HCoV-229E, respectively. Moreover, the total detection time for the rRT-PCR method was about 2 h, and the detection range was 20–2 × 108 copies/reaction for HCoV-OC43 and 200–2 × 109 copies/reaction for HCoV-229E [39].
Tiveljung-Lindell and coworkers developed an rRT-PCR detection method by TaqMan probe for detection of 15 different respiratory viruses, including HCoV-OC43, HCoV-229E, HCoV-NL63, and HCoV-HKU1. They applied this diagnostic platform to 585 stored nasopharyngeal aspirate samples, which were previously evaluated by immunofluorescence and virus isolation methods. The developed rRT-PCR method identified viruses in 57% of the samples within 4 h, while the previous methods only detected viruses in 37% of the samples [41]. Sanghavi et al. evaluated a multiplex rRT-PCR assay which was using TaqMan probe. They tested 728 clinical respiratory specimens for 19 different viruses such as Flu A, Flu B, HCoV-OC43, and HCoV-229E. These experiments were performed at 2 h and 30 min, and the LOD was 5 × 100.5 TCID50/ml and 5 × 101.5 TCID50/ml for HCoV-OC43 and HCoV-229E, respectively [43].
After MERS-CoV appearance in 2012, Corman and colleagues designed an rRT-PCR assay to detect this coronavirus in 92 respiratory swab, sputum, and endotracheal aspirate samples. The probe type was TaqMan. The assay took about 1 h and 20 min to complete, and was able to detect 3.4 copies/reaction or higher [42]. Then, Croman and partners validated a commercial rRT-PCR kit for MERS-CoV that consisted of two assays targeting upstream of the Envelope gene (upE) and open reading frame (ORF) 1a. The kit was able to detect 5.3 copies/reaction by upE assay and 9.3 copies/reaction by ORF1a assay within about an hour [45].
Lu et al. reported four rRT-PCR assays for MERS-CoV detection within 2 h by using specific sequence probes. The TaqMan-type probes were labeled with 6-FAM and Black Hole Quencher 1 as reporter molecule and quencher, respectively. The linear range of their assays was 5–5 × 107 copies/reaction and 10–108 copies/reaction for N assay and upE assay, respectively. Besides, the detection limit for the different assays was in the range of 1.3 × 10−2–1.3 × 10−3 TCID50 and was less than 10 copies/reaction for all the assays [44]. Furthermore, Chan’s group developed rRT-PCR assays with locked nucleic acid probes, which were targeting leader sequences of coronaviruses. They could detect 10 copies/reaction or higher of MERS-CoV, HCoV-OC43, and HCoV-229E and 5 copies/reaction or higher of HCoV-NL63 and HCoV-HKU1. In addition, the LOD values for viral RNA were 5.62 × 10−2, 5.00 × 10−2, and 3.16 × 10−3 TCID50/ml for MERS-CoV, HCoV-229E, and HCoV-OC43, respectively [46].
Hashemzadeh et al. reported a dual TaqMan-based rRT-PCR assay for MERS-CoV detection, which was designed for upE and ORF1b targeting. They evaluated the assay for spiked RNA solutions, and it was able to detect 10 copies/reaction of RNA in about 1 h [47]. Besides, Noh and colleagues developed an rRT-PCR assay for simultaneous detection of SARS-CoV and MERS-CoV and related bat coronaviruses. For this purpose, they used different fluorescent dyes in the probes (HEX and FAM). In the duplex experiment (both coronaviruses detection template), the LOD was 50 ng/ml for MERS-CoV while it was only 1 ng/ml in the single detection template [48].
After COVID-19 outbreak, Corman and coworkers developed an rRT-PCR assay for the detection of SARS-CoV-2 in 2020. This TaqMan-based assay was used for SARS-CoV detection either, and the LOD of the assay was 2.9 and 3.2 copies/reaction for SARS-CoV-2 and SARS-CoV, respectively [49]. Pfefferle and teammates performed another study for SARS-CoV-2 by using the automated rRT-PCR method. They evaluated their assay for 88 swab samples and the LOD was 275.7 copies/reaction. Besides, the total detection time was decreased to about an hour [50]. Also, Hirotsu et al. designed a double quencher probe for the rRT-PCR method. They could detect 10 copies/reaction of SARS-CoV-2 in about 30 min in the positive plasmid controls, containing the complete N gene [51].
It should be noted that the recommended WHO protocol for SARS-CoV-2 detection is the rRT-PCR method with TaqMan probe which has been widely used for COVID-19 diagnosis worldwide. This assay is performed more than 1,000,000 tests per day for the time being [87, 88].
Currently, besides the WHO’s suggested protocol, several other commercial rRT-PCR assays with the United States Food and Drug Association (FDA) approval are being employed to clinically diagnose COVID-19. The AllplexTM 2019-nCoV diagnostic kit (Seegene Inc., Seoul, South Korea), for instance, is an assay that concurrently detects three SARS-CoV-2-associated genes in a single tube, i.e., E, N, and RNA-dependent RNA polymerase (RdRP), using FAM, Quasar® 670, and CAL Fluor® Red 610 fluorophore probes, respectively. Human nasopharyngeal, oropharyngeal, or anterior nasal swabs, mid-turbinate, and sputum specimens from COVID-19 suspects may be used as sources of these nucleic acids, and the results will be available within 1 h and 50 min after RNA extraction. The assay’s LOD has been confirmed as 1250 or 4167 copies/ml, depending on whether a CFX96TM or CFX96 TouchTM Real-Time PCR Detection System is utilized for amplification, respectively [52, 53].
Moreover, GeneFinderTM COVID-19 Plus RealAmp kit (Osang Healthcare Co., Ltd., South Korea) is a one-step rRT-PCR-based assay that detects the three previously mentioned SARS-CoV-2-related genes in nasopharyngeal, oropharyngeal, nasal, or mid-turbinate nasal swab samples, bronchoalveolar lavage fluid, or sputum specimens within roughly 2 h in a single tube. The E, N, and RdRP genes are targeted by probes tagged with Texas Red, JOE, and FAM fluorescent reporter dyes, respectively. This diagnostic kit has an established LOD of 500 copies/ml. Therefore, it is much more sensitive than the AllplexTM 2019-nCoV assay [54, 55].
Additionally, cobas® SARS-CoV-2 test (Roche, Basel, Switzerland) is another rRT-PCR assay that simultaneously targets and detects two SARS-CoV-2-associated nucleic acids in nasopharyngeal or oropharyngeal swab specimens, producing virtually 96 results within 3 h, an overall of 384 or 1056 results for the cobas® 6800 or 8800 System in approximately 8 h, respectively. The LOD of this detection kit has been reported as 0.009 and 0.003 TCID50/ml for ORF1a and E genes, respectively [56, 57].
Furthermore, the RealStar® SARS-CoV-2 RT-PCR kit (altona Diagnostics GmbH, Hamburg, Germany), a real-time assay, targets the E and S genes’ nucleic acid sequences using probes labeled with FAM and Cy5 fluorophores, respectively. These RNA sequences are extracted from COVID-19 suspects’ samples, including nasopharyngeal, oropharyngeal, anterior nasal, or mid-turbinate nasal swabs, nasal washes or aspirates, through AltoStar® Automation System AM16, then purified by the AltoStar® Purification Kit 1.5, and further amplified and identified by CFX96TM or CFX96 TouchTM Real-Time PCR Detection System. This assay has an LOD of 625 copies/ml, demonstrating its higher sensitivity compared with the suggested WHO protocol with an LOD of 1250 copies/ml [58, 59].
Another rRT-PCR-based COVID-19 diagnostic kit is the Xpert® Xpress SARS-CoV-2 test (Cepheid Inc., Sunnyvale, California, USA), which detects E and N2 nucleic acid targets in nasopharyngeal, oropharyngeal, nasal, or mid-turbinate swabs, nasal washes or aspirates from COVID-19 suspects. The rapid assay takes roughly 30 min to generate the results and has an LOD of 250 copies/ml for SARS-CoV-2 reference material or 0.0100 plaque-forming units (PFU)/ml for live virus molecules [60, 61].
Isothermal amplification-based methods
DNA amplification techniques are beneficial for the identification of genetic disorders or infectious diseases. There are different nucleic acid amplification methods other than PCR such as nucleic acid sequence-based amplification (NASBA), transcription-mediated amplification (TMA), loop-mediated isothermal amplification (LAMP), isothermal multiple displacement amplification (IMDA), strand displacement amplification (SDA), signal-mediated amplification of RNA technology (SMART), and helicase-dependent amplification (HDA). Among these methods, LAMP is the second most common after PCR, especially for the detection purposes. LAMP technique requires 4 (or 6) primers which divide the target sequence to six regions (3 regions in the forward section and 3 regions in the backward section). Therefore, the design of primers and their conflict prevention are more challenging compared to the PCR method, which only needs two primers (forward and backward). On the other hand, the LAMP process is carried out in an isothermal condition and it is a cheaper and faster method. Moreover, several researches reported the application of LAMP method for coronaviruses detection [28, 29, 89].
The schematic illustration of LAMP process is represented in Fig. 4. Double-stranded DNA must be converted to single-stranded DNA at the first step. For the left strand in the figure, the forward internal primer (FIP) and F3 primer are hybridized to target, and DNA polymerase is implemented to form a new strand from free nucleotides in the mixture. The newly formed strand is separated due to the zip-like behavior of F3 primer. These steps are repeated in the backward section by backward internal primer and B3 primer. Then, the two ends of the final strand are hybridized to the inner sections to form a loop-shaped strand and the amplification is continued exponentially. For the RNA amplification, reverse transcription step must be done before the LAMP process, which is known as the RT-LAMP method. Detection of the amplicon is usually performed by observing the precipitates of magnesium pyrophosphate by-product, DNA-binding dyes, gel electrophoresis, or real-time fluorescence [30, 90, 91].

Schematic illustration of LAMP process
The first research on human coronaviruses detection by RT-LAMP method was conducted in 2011. Pyrc et al. developed an RT-LAMP assay for the detection of HCoV-NL63, which was able to detect even 1 copy/reaction in clinical specimens. They used gel electrophoresis for the amplicon identification step, and the detection time of the assay was about 1 h [62].
After MERS-CoV discovery, 5 different researches detected this coronavirus by a rapid RT-LAMP method between 2014 and 2018. The detection time for the reported assays was 30–60 min. Shirato’s group developed an assay by real-time fluorescence detection in 2014 with 1.6 copies/reaction. They reported a new real-time assay in 2018 by using a quenching probe. The new assay’s LOD was 15 copies/reaction while the detection time decreased from 1 h to 30 min. Bhadra et al. presented another real-time fluorescence assay which was able to detect 72 copies/reaction within 30–50 min [63, 64, 67]. Lee et al. used agarose gel electrophoresis for amplification product identification, and their LOD and detection time were 0.4 copies/reaction and 1 h, respectively. Huang and colleagues used a vertical flow visualization strip for final amplicon detection and detected 10 copies/μl within 35 min [65, 66].
Besides, Kim et al. developed a multiplex RT-LAMP for detection of SARS-CoV in synthetic RNA solutions by targeting ORF1b and N genes. They investigated both gel electrophoresis and real-time fluorescent methods for final RNA identification. The LOD and detection time of the assay were 104 copies/reaction and 20–25 min, respectively [68].
There are several studies for the rapid diagnosis of COVID-19 employing the RT-LAMP technique. Yan et al. designed primer sets for ORF1ab and S genes for COVID-19 diagnosis within 20–30 min in 130 nasopharyngeal swab and bronchoalveolar lavage fluid samples. The LOD values were 20 and 200 copies/reaction for ORF1ab assay and S gene assay, respectively [69]. Moreover, Yang and coworkers presented an RT-LAMP assay by targeting ORF1ab, E, and N genes to achieve high sensitivity and specificity. After investigation of 208 nasopharyngeal swab specimens, the sensitivity was similar to RT-PCR as the standard assay and the specificity was 99%. The LOD and the detection time of this assay were 5 copies/reaction and 30 min, respectively. It should be noted that in both mentioned assays, calcein dye was used for the identification step [28].
Zhang and teammates targeted ORF1a and N genes, and they could detect SARS-CoV-2 after about 1 h by a simple colorimetric identification. They performed real-time measurements either, and the LOD was 120 copies/reaction [70]. Also, Zhu et al. designed primer sets for ORF1a and N genes, and the RT-LAMP product was visualized by nanoparticles-based biosensor for COVID-19 diagnosis. Their tested samples were oropharynx swab specimens, and both the sensitivity and specificity of the assay were 100% comparing to the rRT-PCR results. The total detection time from sample collection to final identification was 1 h, and the LOD was 12 copies/reaction [71]. Furthermore, Butt et al. reported an RT-LAMP assay by ORF1a and N genes targeting and simple colorimetric identification. They tested the assay on 70 nasopharyngeal swabs and the sensitivity and specificity of the test were 95% and 100%, respectively, when compared to the rRT-PCR results [72].
In the other colorimetric RT-LAMP assay, Lu et al. designed 6 primer sets and could detect 3 copies/reaction of SARS-CoV-2 in about 50 min [73]. Broughton et al. reported the next RT-LAMP based assay, which used the clustered regularly interspaced short palindromic repeats (CRISPR) method for the detection step by lateral flow strip. Their assay was able to detect 1.2 copies/reaction of SARS-CoV-2 within 40 min. It should be noted that CRISPR is a gene-editing technique that could detect a specified sequence and edit it. Recently, this technique is applied for the detection of the specified nucleic acid sequences. [74]
The next isothermal nucleic acid amplification method is recombinase polymerase amplification (RPA), which is working on body temperature (37–42 °C). This method employs three enzymes. A recombinase is pairing to oligonucleotide primers with a homologous sequence in DNA. A single-stranded DNA-binding protein prevents the primers from displacement by binding to displaced DNA strands. The third enzyme, strand-displacing polymerase, synthesizes the DNA starting from the primer-DNA binding point. So, on the presence of forwarding and backward primers, the amplification process goes on like PCR. For RNA amplification, the reverse transcription step must be carried out first [92, 93].
El Wahed et al. reported an RT-RPA-based assay for detection of MERS-CoV NC gene fragment, while a fluorescent tube-scanner was used for amplification detection. The detection time and LOD were 10 min and 10 copies/reaction, respectively [75].
Afterward, an all-in-one assay is developed by Koo and coworkers by using the RT-RPA technique. This device, which was based on an arch-shaped direct amplification silicon microring resonator (SMR) platform, comprising the immobilized recombinase polymerase amplification enzymes and primers specific to the targets, allowed for the high-speed simultaneous analysis of multiple viruses, including MERS-CoV and HCoV-OC43. Instead of employing the hybridization reaction concept, this assay utilized long primers at high concentrations to produce an arch on the SMR platform, which then allowed for label-free detection of viral RNA sequences in a real-time manner. This assay could detect pathogens within 20 min and observed a detection limit of 10 RNA copies/reaction for both coronaviruses [76, 77].
Curti and colleagues reported another RT-RPA assay combined with the CRISPR technique for SARS-CoV-2 detection. They applied both lateral flow and ssDNA fluorescent reporter for the final visualization of the result. The LOD and the detection time of their method were 10 copies/μl and 1 h, respectively [78]. Ding et al. designed an all-in-one RT-RPA assay that used the CRISPR technique with ssDNA fluorescent reporter for the detection step of COVID-19 and HIV diagnosis. The assay progress took about 40 min to complete, and it could detect 1.3 copies/reaction of SARS-CoV-2 [79].
Insulated isothermal polymerase chain reaction (iiPCR) is another amplification method, which was used by Go et al. for MERS-CoV diagnosis. Ralyeigh-Benard convection is the base of this method. There is a thermal gradient in the amplification reactor, but each part of the reactor works at a constant temperature. In this method, the positions of the molecules are changed during the PCR progress instead of changing the whole reactor temperature. Go and coworkers used TaqMan probes for the rapid detection of MERS-CoV in tissue culture fluid and sputum specimens by RT-iiPCR method. The detection time of the assay was about 1.5 h, and its LOD was 0.37 PFU/ml, which was equal to less than 10 copies/reaction [80, 94].
Biosensors for viral RNA detection
RNA-based biosensors are highly promising substitutes for the conventional RT-PCR diagnostic techniques due to their simplicity, cost-effectiveness, rapid detection, and high sensitivity and specificity; particularly, since they can eliminate the need for RNA amplification [95,96,97,98]. In the field of coronaviruses diagnosis, most assays make use of the binding of synthetic single-stranded DNA probes to the target viral RNA or oligonucleotide sequences through hybridization reactions, which result in the formation of double-stranded nucleic acids [99]. Free or immobilized DNA probes were tagged with transducing elements in advance or become subjected to them after hybridizing with the targets. Electrochemical [100,101,102,103] and optical agents [24, 37, 77, 104,105,106] are examples of transducers that have been employed in these gene-based sensors to produce measurable signals corresponding to the presence or absence of viral RNA. Figure 5 presents a schematic of the principle underlying of these biosensors. Table 2 summarizes the RNA-based biosensors developed for the detection of coronaviruses.

RNA-based biosensors for coronaviruses detection based on a the hybridization reaction between DNA probes tagged with transducing elements and viral sequences, or b the reaction between DNA probes and targets, and the subsequent introduction of transducing agents
Electrochemical biosensors
These biosensors are appropriate candidates for amplification-free detection of viral RNA; since they offer minimum sample requirement, simple operation, inexpensiveness, and ultrasensitive rapid real-time detection [108, 109]. In these sensors, biorecognition cues triggered by the hybridization reactions result in assessable impedimetric, potentiometric, chronoamperometric, or voltammetric signals. The most common electrochemical techniques in viral biosensors are cyclic voltammetric (CV), square-wave voltammetry (SWV), chronoamperometry (CA), electrochemical impedance spectroscopy (EIS), and differential pulse voltammetry (DPV) [110, 111]. Various studies have exploited these sensors to detect SARS-CoV-specific 30-mer oligonucleotides. Abad-Valle et al. developed a genosensor for the detection of these oligonucleotide sequences. In this hybridization-based electrochemical assay, thiol-terminated complementary probes for the specific target of viral sequences were immobilized on a sputtered gold film through sulfur-gold interactions. After surface blocking with 1-hexanethiol, the hybridization reaction between the probes and biotinylated target strands took place. Subsequently, the insertion of streptavidin (Strp)-tagged alkaline phosphatase (AP) enabled the electrochemical detection of SARS-related virus; this enzyme hydrolyzed the deposited 3-indoxyl phosphate (3-IP) to soluble indigo carmine, which indirectly led to the identification of the oligonucleotide sequences. The biosensing progress took approximately 2 h to complete and employed SWV, as the electrochemical measurement technique, which resulted in signals linearly correlating with a concentration range of 0.01–1.01 nM and a detection limit of 6 pM [100]. González-López and Abedul also detected these oligonucleotides in spiked solutions with the same described platform and electrochemical measurement method but within a linear range of 0.1–10 nM and an assay time of 3 h [103].
Additionally, Díaz-González and coworkers constructed two hybridization-based biosensors for the detection of these 30-mer oligonucleotides. In both sensors, screen-printed carbon electrodes (SPCE) were electrochemically pre-oxidized and then modified by cationic polylysine. DNA probes were then immobilized onto the surface of the modified electrodes via electrostatic interactions. One of these biosensors was based on the direct CV detection of indigo carmine, the product of 3-IP hydrolysis in the presence of AP. This sensor had a detection time of 3 h and 25 min with a detection limit of 8 pM. In another sensor, target oligonucleotides labeled with Au (I) complex were detected by CA through the hydrogen evolution reaction catalyzed by this label. A detection limit of 0.5 nM in a remarkably lower time-frame of 90 min were observed for this assay [102].
Moreover, Martínez-Paredes and colleagues reported an electrochemical hybridization-based genosensor incorporating an SPCE modified with gold nanoparticles. Thiolated single-stranded DNA probes complementary to the 30-mer SARS-CoV-specific oligonucleotides were immobilized onto the modified electrode. After the blocking step with casein, a hybridization reaction occurred with the insertion of biotinylated target oligonucleotides. The targets were then labeled with AP through biotin-Strp binding. After the addition of a 3-IP and silver nitrate solution, 3-IP produced a compound that reduced the silver ions to metallic silver and deposited them on the AP labels, which were then oxidized and measured by anodic stripping voltammetry. The usage of gold NPs and the stripping voltammetry of silver ions improved the sensitivity of this assay by several orders of magnitude, when compared to usage of gold films and direct voltammetric measurement of indigo carmine. Upon using CV, a linear range and sensitivity of 2.5–50 pM and 1.76 μA/pM were reported for this assay, respectively [101].
Optical biosensors
These biosensors detect the biorecognition process between the capture probes and target analytes without a direct electrical connection. Optical detection has enabled the construction of cost-effective, easy-to-operate sensors for rapid and sensitive analysis of pathogens, with the option of high-throughput screening [110, 112]. The optical biosensors designed for detecting coronaviruses-specific RNA sequences are based on SPR [24, 37], fluorescence [104], SMR [77], and colorimetry [105,106,107].
Several conducted studies incorporated optical platforms for coronaviruses RNA detection, but they were not sensitive enough to eliminate the RNA amplification step. In order to decrease total detection time, they have been designed all-in-one instruments. As mentioned in the previous section, Shi and coworkers reported an optical biosensor based on SPR coupling with RT-PCR step, which detected SARS-CoV and eight other respiratory viruses. Also, Koo et al. presented an arch-shaped direct amplification SMR platform for simultaneous detection of MERS-CoV and HCoV-OC43 and some other viruses [37, 77]. Furthermore, In an attempt to create all-in-one direct RNA amplification and sensing platforms, Jung et al. designed a microfluidic chip, based on DNA hydrogel development by isothermal amplification of complementary target sequences (DhITACT-TR) for rapid and robust detection of MERS-related coronavirus RNA in pseudo-serum specimens. The optical device provided an observable result by the naked eye or fluorescent ultraviolet light. In comparison to the laborious conventional RT-PCR procedure, this 30-min assay only required the injection of the extracted RNA into the chip, which led to the hybridization reaction between first and second generation of amplified DNA strands. This method could detect as low as 0.1 pM of pathogenic targets [104].
The next step was to design ultrasensitive biosensors that would eliminate the need for RNA amplification and provide straightforward and rapid detection platforms. Li and Rothberg proposed a colorimetric assay for SARS-CoV oligonucleotide sequence detection. This sensor exploited the different inclinations single-stranded and hybridized DNA molecules possessed for electrostatic adsorption on colloidal gold NPs. They demonstrated that in contrast to the hybridized oligonucleotides, single-stranded DNA molecules stabilized the NPs and prevented them from aggregation after salt addition. Thus, this biosensor detected the change of color prompted by the NPs aggregation, as a result of the hybridization reaction. The assay progress took about 5 min and enabled visual detection for < 100 fmol of the targets [107].
Furthermore, Teengam and coworkers developed a paper-based colorimetric genosensor, which operated based on the citrate-stabilized silver NPs aggregation promoted by the presence of positively charged pyrrolidinyl peptide nucleic acids (PNA). PNA were used as substitutes for RNA probes, due to their stability, ease of synthesis, and efficient hybridization with the target sequences. In the presence of synthetic MERS-CoV oligonucleotides, PNA were hybridized with the targets and produced anionic double-stranded molecules, which led to the dispersion of the silver particles, due to the electrostatic repulsion, and generated a visual color change. A linear concentration range of 20–100 nM was observed for spiked target sequence solutions with a detection limit of 1.53 nM [106]. Another work in the field of MERS-related coronavirus detection is the study carried out by Kim et al. They designed a colorimetric biosensor that detected MERS-CoV-specific oligonucleotides. In the absence of targets, the two designed thiol-terminated probes interconnected together through disulfide bond formation and failed to shield the citrate-capped gold NPs from positively charged electrolyte-induced particle aggregation. However, in the presence of the targets, disulfide-induced self-assembly of the targets and the two probes took place. These products acted as shields for the NPs, due to the sulfur-gold covalent bonds formed on the particles’ surface, resulting in a substantial color change visible to the naked eye. The 10-min assay had a detection limit of 1 nM for 30 bp MERS-CoV oligonucleotides [105].
In the context of COVID-19 diagnosis, Qiu et al. proposed an optical biosensor exploiting localized surface plasmon resonance (LSPR) and plasmonic photothermal effect. In this assay, the SARS-CoV-2 oligonucleotides were hybridized with the immobilized DNA probes on the gold nanoislands-modified platform. The photothermal heat triggered by the illumination of the substrate at gold NPs’ LPSR frequency was able to increase the hybridization temperature and provide an ultrasensitive assay. The designed biosensor detected as low as 0.22 pM within a relatively short time of 14 min [24].
Whole virus or viral proteins detection
In addition to coronaviruses-related disease diagnosis through CT imaging, RT-PCR, direct RNA biosensing, and host antibody detection, an alternative strategy is to identify the viral proteins or whole virus in collected samples. Spike and N proteins are two of the four structural proteins in coronaviruses [113]. Spike protein is one of the membrane proteins that makes up the viral envelope. This 180 kDa glycoprotein comprises S1 and S2 subunits. The S1 subunit, an extremely immunogenic antigen, is the main target for neutralizing antibodies, since it encompasses a receptor-binding domain. Therefore, S1-specific antibodies have been studied throughout the years as bioreceptors for whole virus detection [114, 115]. N protein, a 40 kDa phosphoprotein encapsulating the viral RNA, can be directly detected in serum, nasopharyngeal aspirate, and urine samples within the first 10 days of infection, with a positive detection rate higher than that of the RT-PCR analyses [116]. Its detectable characteristic is due to its relatively large size and using sandwich-like immunoassays for its detection and also, the high immunogenicity that it exhibits during this period [117]. Albeit, studies have shown that the sensitivity of the N protein detection reduces to less than 30% when more than 11 days have elapsed since the onset of the viral invasion [118]. Hence, there is still a substantial need for developing ultrasensitive assays to detect this viral protein [119]. Consequently, since the outbreak of coronaviruses in 2002, efforts have been made to produce assays with high sensitivity to detect viral N proteins or whole coronaviruses by targeting their spike structural proteins.
Figure 6a depicts a schematic of whole virus-based assays designed for coronaviruses detection. In most cases, immobilized antibodies specific to spike proteins are used as bioreceptors to capture the virus molecules. Subsequently, label-free or label-based detection is carried out to analyze the captured analytes. In addition to the conventional enzyme-linked immunosorbent assay (ELISA) technique [120], electrochemical [121, 122], electrical [27, 123], and mass-based biosensors [124], have been developed to detect coronaviruses via their spike proteins. As illustrated in Fig. 6b, the detection of viral N proteins has almost the same concept as identifying whole virus molecules but with slight differences. Most biosensors designed for this cause have utilized antibodies specific to N proteins as the capture probes. After the capturing step, label-free or label-based detection has been accomplished by employing optical [125,126,127] and electrochemical transducers [121]. Apart from these biosensors, typical western blotting, ELISA, and lateral flow immunoassay (LFIA) have also been exploited in diagnosing coronaviruses-related diseases through the viral N protein detection [118, 128,129,130,131,132]. However, it is notable that a couple of the proposed biosensors for either whole virus or N protein detection have been incorporated with other bioreceptors or detection probes, which will be further elucidated in the following sections. Table 3 provides a summary of the developed assays for the detection of coronaviruses through whole virus or viral N protein analysis.

The general principle of a whole virus- and b viral N protein-based assays for coronaviruses identification by label-free or label-based detection. In both cases, antibodies are used as capture and detection probes, if label-based detection is employed
Conventional detection methods
Traditional assays, including ELISA, LFIA, and western blotting, have been proposed for the detection of whole coronaviruses via their spike membrane proteins, or viral N proteins.
Whole virus detection
ELISA technique has been employed in whole virus detection by targeting its spike proteins. In 2013, Sunwoo and coworkers reported an assay for SARS-related coronavirus detection, which exploited anti-S1 monoclonal capture antibodies, as well as HRP-labeled bi-specific monoclonal detection antibodies to detect the S1 subunits of spike proteins by the addition of 3,3′,5,5′-Tetramethylbenzidine (TMB) chromogenic substrate. This method detected as low as 19 ng/ml in approximately 2 h [120].
Viral N protein detection
Conventional detection techniques such as western blotting, ELISA, and LFIA have been utilized to detect coronaviruses N proteins in spiked or real clinical samples. For instance, Che and colleagues employed the western blot analytical method to detect recombinant His6-tagged viral N proteins specific to SARS-CoV, HCoV-OC43, and -229E using anti-His monoclonal antibodies [132]. The popular ELISA technique has also been extensively used for SARS- and MERS-CoV N protein detection. In a study performed by Lau and coworkers, an ELISA was designed to identify SARS-specific N proteins in nasopharyngeal aspirate and serum samples within 3 h and 31 min. The proteins were first captured by guinea pig anti-N protein antibodies coated on immunoplates. Then, rabbit anti-N protein antibodies were introduced to form the sandwich-like structures. After the addition of HRP-tagged goat anti-rabbit antibodies, detection was carried out by incubating the plates with TMB substrate. The method observed detection limits of 2.5 ng/ml and 0.4 ng/ml for nasopharyngeal aspirate and urine specimens, respectively [118]. In another work, Kammila et al. developed a rapid immunoswab assay for SARS-CoV-related N protein detection. This assay was a pseudo-ELISA performed on the surface of calcium-alginate swabs used for collecting the nasopharyngeal aspirate samples. After the capture of N proteins by the antibodies immobilized on the swab surface and incubation with Immunoglobulin Y (IgY), HRP-tagged rabbit anti-chicken IgY antibodies were added, followed by incubation with TMB. The 90-min assay had a detection limit of 0.5 pg/swab or 10 pg/ml for NA samples [129].
Moreover, in 2015, Chen’s group proposed an ELISA for MERS-CoV N protein detection and quantification. Two anti-MERS-CoV recombinant N protein antibodies were used as the capture and detection probes. The assay principle was similar to prior studies. The designed ELISA took about 1 h and 12 min and detected as low as 1 ng/ml of the analytes in spiked recombinant N protein solutions [128]. Also, in 2016, they designed an LFIA for MERS disease diagnosis via N protein detection using two specific monoclonal antibodies against MERS-CoV recombinant N proteins. The 30-min rapid detection assay had a sensitivity and detection limit of 81% and 103.7–104.2 TCID50/ml for spiked nasopharyngeal aspirate samples, respectively [130]. Additionally, in 2020, Fung et al. provided a step-by-step protocol for the detection of MERS-CoV using an ELISA. The proposed assay employed two N protein-specific monoclonal antibodies and took 1 h and 12 min to complete [131]. Furthermore, in a recent work, Li and coworkers developed an ELISA to measure the concentration of inactivated SARS-CoV-2 N proteins in vaccines. The 2-h assay, which utilized polyclonal anti-N protein antibodies to form the sandwich-like structures had a detection limit of 100 ng/ml within a concentration range of 200–1600 ng/ml [140].
The first FDA-approved coronavirus-associated antigen detection assay was introduced on the 10th of May, 2020 as a means to rapidly diagnose COVID-19. The Sofia SARS Antigen FIA (Quidel Corp., San Diego, California, USA) is a fluorescence-based sandwich LFIA that enables the qualitative detection of the SARS-CoV and SARS-CoV-2 N proteins from nasopharyngeal and nasal swabs within 15 min. Nevertheless, the test cannot distinguish between the two viruses. This assay has observed an LOD of 113 TCID50/ml, with a clinical sensitivity and specificity of 80% and 100%, respectively [133, 134].
Biosensors
As clarified earlier, on top of the use of conventional techniques for coronaviruses-related disease diagnosis, biosensors have become emerging platforms for whole virus or viral N protein detection. According to the related studies conducted to date, the developed biosensors were either electrochemical, electrical, optical, or mass-based.
Electrochemical and electrical biosensors
The merits of electrochemical biosensors were discussed earlier in Electrochemical biosensors section. These sensors are also ideal candidates for whole virus or viral protein detection. Layqah and Eissa proposed an electrochemical immunosensor for the detection of MERS-CoV and HCoV-OC43 using a panel of gold NPs-modified carbon working electrodes. The S1 subunits of spike proteins and N proteins were the target analytes for MERS whole virus and HCoV-OC43 protein detection, respectively. The target molecules competed with the immobilized MERS-CoV S1 subunits or HCoV-OC43 N proteins, for specific antibodies further added to the platform. Potassium ferrocyanide/potassium ferricyanide complex was used as the redox probe, and label-free electrochemical detection was achieved by SWV. The 20-min assay observed signals linearly correlating with concentration ranges of 1 pg/ml–100 ng/ml and 10 pg/ml–10 μg/ml, and also, detection limits of 1.04 pg/ml and 0.4 pg/ml for MERS-CoV and HCoV-OC43, respectively [121].
Furthermore, in a recent study regarding the detection of SARS-CoV-2, Mahari and teammates designed two label-free electrochemical biosensors that identified the SARS-CoV-2 virus in spiked saliva samples via its spike proteins. Both biosensors employed antibodies as capture probes for these membrane proteins. The first sensor, a potentiostat-based device, used a fluorine-doped tin oxide working electrode modified with gold NPs. After investigating its analytical performance using the DPV technique, it was shown that it could detect SARS-CoV-2 in the concentration range of 1 fM–1 μM, with an LOD of 10 fM. The second one utilized an SPCE with an in-house built electrochemical device called eCovSens, which correlated voltage with concentration. This device could also detect as low as 10 fM of spike proteins, in a relatively short assay time of 1 min [122].
Field-effect transistors (FET) are electrical biosensors consisting of source and drain units separated by a channel, all typically fabricated on a silicon/silicon dioxide substrate. Their simplicity and low-cost fabrication, as well as ultrasensitive, label-free, and rapid real-time detection, have made them highly attractive biosensing platforms [141]. In 2009, Ishikawa’s group developed a FET-based biosensor for the detection of SARS-CoV N proteins. Instead of using antibodies against these analytes, they employed fibronectin, as antibody-mimicking polypeptides. The sensor had gold source and drain electrodes, which were linked by an indium oxide nanowire. The device detected viral N proteins in only 10 min [135]. In 2010, this group reported another FET-based sensor for SARS-CoV N protein detection. The platform was almost the same as the previous one, except that it had single-walled carbon nanotubes as the channel linking the gold electrodes. The 10-min assay could detect between 5 and 50 nM of spiked N protein solutions [136].
Due to the capability of FET-based biosensors to detect minute amounts of analytes with ultrahigh sensitivity, they have recently been thoroughly utilized in developing assays for SARS-CoV-2-related whole virus detection via spike membrane proteins. For instance, Seo and colleagues designed a label-free FET-based device with gold source and drain electrodes, as well as a graphene sheet channel with anti-spike protein antibodies immobilized onto it. The device could detect as low as 100 fg/ml of the analytes in the clinical transport medium used for nasopharyngeal aspirate swabs [27]. Furthermore, Zhang et al. proposed another graphene FET-based biosensor that could detect COVID-19-related virus via its spike proteins in about 2 min, again by employing spike protein-specific antibodies. A 0.2 pM detection limit was observed for the assay [123].
Optical biosensors
The benefits of optical biosensors were previously explained in Optical biosensors section. This section reviews the optical biosensors reported for coronavirus-related N protein detection; including chemiluminescent [138], fluorescent [126, 138, 139], SPR-based [137], and localized surface plasmon-coupled fluorescence (LSPCF) fiber-optic biosensors [125, 127].
Fluorescence-based detection has been investigated as a means to detect viral N proteins. Ahn and coworkers developed two methods for the detection of SARS-CoV N protein, a chemiluminescent assay and a fluorescent one. Both assays employed aptamers specific to the proteins as bioreceptors, and polyclonal anti-N protein antibodies to create the sandwich-like structures. The 3-h chemiluminescent technique, then used AP-conjugated anti-immunoglobulin G (IgG) antibodies, followed by the addition of disodium 3-(4-methoxyspiro {1,2-dioxetane-3,2′-(5′-chloro) tricyclo [3.3.1.13,7] decan}-4-yl) phenyl phosphate as the chemiluminescent substrate, and observed a detection limit of 20 pg/ml. In comparison, the 4-h fluorescent assay exploited fluorescein isothiocyanate (FITC)-labeled anti-IgG and detected as low as 2 pg/ml [138]. In another study, Roh and Jo designed a chip that detected SARS-CoV N proteins in only an hour with a detection limit of 0.1 pg/ml. The target proteins were immobilized on the chip via ProLinker™ capture probes, then quantum dots-labeled specific aptamers bound to the targets, and fluorescence intensity was evaluated using confocal microscopy [139]. In a recent study, Diao and colleagues proposed a fluorescent immunochromatographic strip assay that detected the N proteins in nasopharyngeal aspirate and urine samples of COVID-19 patients that took only 10 min with a sensitivity of 68–98%. The sealed strip comprised sample and conjugate pads, a nitrocellulose membrane with test and control lines on its surface, and also an adsorption pad. The conjugate pad contained europium (III)-labeled monoclonal anti-N protein (M4) or IgG antibodies. Monoclonal anti-N protein (M1) and anti-IgG antibodies were immobilized onto the test and control lines, respectively. Due to the capillary effect, N proteins present in a given sample bounded to the labeled M4 or IgG antibodies located on the conjugate pad and, then, were captured by the M1 or anti-IgG antibodies on the test and control lines, respectively; leading to fluorescent-colored lines. The results were further assessed by an immunofluorescence analyzer [126].
SPR-based biosensors are promising platforms for the detection of pathogens, including coronavirus-specific N proteins, due to their label-free real-time sensing characteristic. Yang et al. developed a Strp-coated chip to study the RNA-N protein interactions related to SARS-CoV. Immobilized biotinylated DNA oligonucleotides underwent a hybridization reaction with synthetic RNA probes specific to SARS-CoV N proteins, and then, after the introduction of the target proteins, the probe-target interactions were investigated by SPR with high sensitivity [137]. Moreover, Chang and Huang’s research group designed two biosensors incorporating LSPCF-based fiber optics for SARS-CoV-specific N protein detection. Each sensor utilized a polymethyl methacrylate fiber as the substrate and exploited the sandwich-like structure by using two monoclonal anti-N protein antibodies as the capture and detection probes. The fluorophore-labeled detection probes were tagged with protein A molecules conjugated to gold NPs. Evanescent waves, exciting the surface plasmons, enhanced the electromagnetic field surrounding the NPs and led to the excitation of fluorophores. Both sensors could detect SARS-CoV N protein within a range of 0.1 pg/ml–1 ng/ml in serum samples. An assay time of 2 h was reported for one of the sensors [125, 127].
Mass-sensitive biosensors
Mass-sensitive biosensors provide label-free detection of pathogens, by directly altering the frequency during the interaction between the analyte and bioreceptor. The most prominent gravimetric sensors developed for pathogenic detection have incorporated quartz crystal microbalance (QCM) as the transducing agent [142]. Zuo and coworkers developed a piezoelectric QCM-based biosensor for the detection of SARS-CoV molecules in spiked sputum specimens. This device enabled the label-free detection of viral particles by the immobilized capture antibodies. The sensor could detect and analyze the presence of the virus in 17 min, within a concentration range of 1–4 μg/ml, and a detection limit of 0.6 μg/ml [124].
Conclusion and future perspectives
The detection methods for all the seven identified human coronaviruses have been reviewed in this article. Due to the significant false-negative results for COVID-19 diagnosis by current methods and non-specific clinical symptoms, especially in early stages, searching for a reliable diagnostic technique will be continued. Also, it will be more challenging if simple usage, low detection time, high sensitivity and specificity, and cost-effectiveness concerns are considered. In the other words, As SARS-CoV-2 is one of the most contagious viruses, we need some simple and portable (or easy to install everywhere) techniques instead of these heavy technical platforms for effective fight against the COVID-19 pandemic.
So far, WHO recommended protocol is based on the rRT-PCR technique. This is an expensive method having a detection time of more than 2 h; nevertheless, the sensitivity is not high enough. RT-LAMP and RT-RPA techniques could be a suitable substitute for RT-PCR because of their similar results to those of RT-PCR, simpler usage, and lower cost, as well as faster detection.
Furthermore, working on biosensors for direct viral RNA detection without amplification is encouraged to attain a cost-effective, rapid, and user-friendly method. SWV- and EIS-based electrochemical and SPR-based optical biosensors are suggested for this purpose.
References
Harapan H, Itoh N, Yufika A, Winardi W, Keam S, Te H et al (2020) Coronavirus disease 2019 (COVID-19): a literature review. J Infect Public Health. https://doi.org/10.1016/j.jiph.2020.03.019
Maier HJ, Bickerton E, Britton P (2015) Coronaviruses: methods and protocols. Springer, Berlin. https://doi.org/10.1007/978-1-4939-2438-7
Guarner J (2020) Three emerging coronaviruses in two decades: the story of SARS, MERS, and now COVID-19. Am J Clin Pathol 153:420–421. https://doi.org/10.1093/ajcp/aqaa029
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423. https://doi.org/10.1002/jmv.25681
Estola T (1970) Coronaviruses, a new group of animal RNA viruses. Avian Dis 14:330–336
Weiss SR (2020) Forty years with coronaviruses. J Exp Med 217. https://doi.org/10.1084/jem.20200537
Kahn JS, McIntosh K (2005) History and recent advances in coronavirus discovery. Pediatr Infect Dis J 24:S223–S227. https://doi.org/10.1097/01.inf.0000188166.17324.60
Breslin JJ, Mørk I, Smith MK, Vogel LK, Hemmila EM, Bonavia A et al (2003) Human coronavirus 229E: receptor binding domain and neutralization by soluble receptor at 37°C. J Virol 77:4435–4438. https://doi.org/10.1128/jvi.77.7.4435-4438.2003
St-Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ (2004) Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol 78:8824–8834. https://doi.org/10.1128/jvi.78.16.8824-8834.2004
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP et al (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science (80- ) 300:1394–1399. https://doi.org/10.1126/science.1085952
Kuiken T, Fouchier RAM, Schutten M, Rimmelzwaan GF, Van Amerongen G, Van Riel D et al (2003) Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270. https://doi.org/10.1016/S0140-6736(03)13967-0
Chiu SS, Hung Chan K, Wing Chu K, Kwan SW, Guan Y, Man Poon LL et al (2005) Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin Infect Dis 40:1721–1729. https://doi.org/10.1086/430301
Woo PCY, Lau SKP, Yip CCY, Huang Y, Yuen KY (2009) More and more coronaviruses: human coronavirus HKU1. Viruses 1:57–71. https://doi.org/10.3390/v1010057
Van Der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJM, Wolthers KC et al (2004) Identification of a new human coronavirus. Nat Med 10:368–373. https://doi.org/10.1038/nm1024
Chan JFW, Lau SKP, To KKW, Cheng VCC, Woo PCY, Yue KY (2015) Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522. https://doi.org/10.1128/CMR.00102-14
World Health Organization. MERS situation update, January 2020 [Internet]. 2020;doi: http://www.emro.who.int/pandemic-epidemic-diseases/mers-cov/mers-situation-update-january-2020.html
Chang L, Yan Y, Wang L (2020) Coronavirus disease 2019: coronaviruses and blood safety. Transfus Med Rev. https://doi.org/10.1016/j.tmrv.2020.02.003
Wang L, Wang Y, Ye D, Liu Q (2020) Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2020.105948
World Health Organization (2020) Coronavirus disease 2019 (COVID-19): situation report, 187
World Health Organization (2020) Coronavirus disease 2019 (COVID-19): situation report, 162
World Health Organization (2020) Coronavirus disease 2019 (COVID-19): situation report, 133
World Health Organization (2020) Laboratory testing for 2019 novel coronavirus (2019-nCoV) in suspected human cases
Zhang W, Du RH, Li B, Zheng XS, Yang X, Lou HB et al (2020) Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect 9:386–389. https://doi.org/10.1080/22221751.2020.1729071
Qiu G, Gai Z, Tao Y, Schmitt J, Kullak-Ublick GA, Wang J (2020) Dual-functional plasmonic photothermal biosensors for highly accurate severe acute respiratory syndrome coronavirus 2 detection. ACS Nano. https://doi.org/10.1021/acsnano.0c02439
Fang Y, Zhang H, Xie J, Lin M, Ying L, Pang P, et al. (2020) Sensitivity of chest CT for COVID-19: comparison to RT-PCR. Radiology ;200432
Zu ZY, Jiang M Di, Xu PP, Chen W, Ni QQ, Lu GM, et al. (2020) Coronavirus disease 2019 (COVID-19): a perspective from China. Radiology ;200490. doi: https://doi.org/10.1148/radiol.2020200490
Seo G, Lee G, Kim MJ, Baek S-H, Choi M, Ku KB et al (2020) Rapid detection of COVID-19 causative virus (SARS- CoV-2) in human nasopharyngeal swab specimens using field-effect transistor-based biosensor. ACS Nano. https://doi.org/10.1021/acsnano.0c02823
Yang W, Dang X, Wang Q, Xu M, Zhao Q, Zhou Y, et al. (2020) Rapid detection of SARS-CoV-2 using reverse transcription RT-LAMP method. medRxiv
Gill P, Ghaemi A (2008) Nucleic acid isothermal amplification technologies — a review. Nucleosides Nucleotides Nucleic Acids 27:224–243. https://doi.org/10.1080/15257770701845204
Craw P, Balachandran W, Craw P (2012) Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip. https://doi.org/10.1039/c2lc40100b
Jalali M, Zaborowska J, Jalali M (2017) The polymerase chain reaction: PCR, qPCR, and RT-PCR. In: Basic science methods for clinical researchers. Elsevier Inc. p. 1–18.doi: https://doi.org/10.1016/B978-0-12-803077-6.00001-1
Vabret A, Mouthon F, Mourez T, Gouarin S, Petitjean J, Freymuth F (2001) Direct diagnosis of human respiratory coronaviruses 229E and OC43 by the polymerase chain reaction. J Virol Methods 97:59–66
Vallet S, Gagneur A, Talbot PJ, Legrand MC, Sizun J, Picard B (2004) Detection of human coronavirus 229E in nasal specimens in large scale studies using an RT-PCR hybridization assay. Mol Cell Probes 18:75–80. https://doi.org/10.1016/j.mcp.2003.09.005
Moës E, Vijgen L, Keyaerts E, Zlateva K, Li S, Maes P et al (2005) A novel pancoronavirus RT-PCR assay: frequent detection of human coronavirus NL63 in children hospitalized with respiratory tract infections in Belgium. BMC Infect Dis 5:1–10. https://doi.org/10.1186/1471-2334-5-6
Lam WY, Yeung ACM, Tang JW, Ip M, Chan EWC, Hui M et al (2007) Rapid multiplex nested PCR for detection of respiratory viruses. J Clin Microbiol 45:3631–3640. https://doi.org/10.1128/JCM.00280-07
Thaitrong N, Liu P, Briese T, Lipkin WI, Chiesl TN, Higa Y et al (2010) Integrated capillary electrophoresis microsystem for multiplex analysis of human respiratory viruses. Anal Chem 82:10102–10109. https://doi.org/10.1063/1.3502457.(33)
Shi L, Sun Q, Xu H, Liu C, Zhao C, Xu Y et al (2015) Development of SPR biosensor for simultaneous detection of multiplex. Biomed Mater Eng 26:S2207–S2216. https://doi.org/10.3233/BME-151526
Poon LLM, Wong BWY, Chan KH, Leung CSW, Yuen KY, Guan Y et al (2004) A one step quantitative RT-PCR for detection of SARS coronavirus with an internal control for PCR inhibitors. J Clin Virol 30:214–217. https://doi.org/10.1016/j.jcv.2003.12.007
Vijgen L, Keyaerts E, Moës E, Maes P, Duson G, Van Ranst M (2005) Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol 43:5452–5456. https://doi.org/10.1128/JCM.43.11.5452-5456.2005
Escutenaire S, Mohamed N, Isaksson M, Thorén P, Klingeborn B, Belák S et al (2007) SYBR green real-time reverse transcription-polymerase chain reaction assay for the generic detection of coronaviruses. Arch Virol 152:41–58. https://doi.org/10.1007/s00705-006-0840-x
Tiveljung-Lindell A, Rotzén-Östlund M, Gupta S, Ullstrand R, Grillner L, Zweygberg-Wirgart B et al (2009) Development and implementation of a molecular diagnostic platform for daily rapid detection of 15 respiratory viruses. J Med Virol 81:167–175. https://doi.org/10.1002/jmv.21368
Corman VM, Eckerle I, Bleicker T, Zaki A, Landt O, Eschbach-Bludau M et al (2012) Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Eurosurveillance 17:1–6. https://doi.org/10.2807/ese.17.39.20285-en
Sanghavi SK, Bullotta A, Husain S, Rinaldo CR (2012) Clinical evaluation of multiplex real-time PCR panels for rapid detection of respiratory viral infections. J Med Virol 84:162–169. https://doi.org/10.1002/jmv.22186
Lu X, Whitaker B, Sakthivel SKK, Kamili S, Rose LE, Lowe L et al (2014) Real-time reverse transcription-pcr assay panel for middle east respiratory syndrome coronavirus. J Clin Microbiol 52:67–75. https://doi.org/10.1128/JCM.02533-13
Corman VM, Ölschläger S, Wendtner CM, Drexler JF, Hess M, Drosten C (2014) Performance and clinical validation of the RealStar® MERS-CoV Kit for detection of Middle East respiratory syndrome coronavirus RNA. J Clin Virol 60:168–171. https://doi.org/10.1016/j.jcv.2014.03.012
Chan JFW, Choi GKY, Tsang AKL, Tee KM, Lam HY, Yip CCY et al (2015) Development and evaluation of novel real-time reverse transcription-PCR assays with locked nucleic acid probes targeting leader sequences of human-pathogenic coronaviruses. J Clin Microbiol 53:2722–2726. https://doi.org/10.1128/JCM.01224-15
Hashemzadeh MS, Rasouli R, Zahraei B, Izadi M, Tat M, Saadat SH et al (2016) Development of dual TaqMan based one-step rRT-PCR assay panel for rapid and accurate diagnostic test of MERS-CoV: a novel human coronavirus, ahead of Hajj pilgrimage. Iran Red Crescent Med J 18:1–7. https://doi.org/10.5812/ircmj.23874
Noh JY, Yoon SW, Kim DJ, Lee MS, Kim JH, Na W et al (2017) Simultaneous detection of severe acute respiratory syndrome, Middle East respiratory syndrome, and related bat coronaviruses by real-time reverse transcription PCR. Arch Virol 162:1617–1623. https://doi.org/10.1007/s00705-017-3281-9
Corman V, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK et al (2020) Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance. https://doi.org/10.2807/1560-7917.ES.2020.25.3.2000045
Pfefferle S, Reucher S, Nörz D, Lütgehetmann M (2020) Evaluation of a quantitative RT-PCR assay for the detection of the emerging coronavirus SARS-CoV-2 using a high throughput system. Eurosurveillance 25:2000152. https://doi.org/10.2807/1560-7917.ES.2020.25.9.2000152
Hirotsu Y, Mochizuki H, Omata M (2020) Double-quencher probes improved the detection sensitivity of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by One-Step RT-PCR. medRxiv;0–2. doi: https://doi.org/10.1101/2020.03.17.20037903
Seegene Inc. (2020) Allplex™ 2019-nCoV Assay [Internet];doi: http://www.seegene.com/assays/allplex_2019_ncov_assay#
Seegene Inc. (2020) Allplex™ 2019-nCoV Assay Instructions for Use [Internet]. doi: https://www.fda.gov/media/137178/download
Osang Healthcare Co. (2020) GeneFinder™ COVID-19 Plus RealAmp Kit Instructions for Use [Internet]. doi: https://www.fda.gov/media/137116/download
Osang Healthcare Co. (2020) GeneFinder™ COVID-19 Plus RealAmp Kit [Internet];doi: http://www.osanghc.com/en/products_en/molecular-diagnosis/#
Roche. (2020) cobas® SARS-CoV-2 Qualitative assay for use on the cobas® 6800/8800 systems, for in vitro diagnostic use [Internet]. doi: https://www.who.int/diagnostics_laboratory/eul_0504-046-00_cobas_sars_cov2_qualitative_assay_ifu.pdf?ua=1
Roche. (2020) cobas® SARS-CoV-2 Test [Internet];doi: https://diagnostics.roche.com/global/en/products/params/cobas-sars-cov-2-test.html
altona Diagnostics GmbH (2020) RealStar® SARS-CoV-2 RT-PCR Kit U.S. Instructions for Use;1–52. doi: https://www.fda.gov/media/137252/download
Visseaux B, Le Hingrat Q, Collin G, Ferré V, Storto A, Ichou H et al (2020) Evaluation of the RealStar® SARS-CoV-2 RT-PCR Kit RUO performances and limit of detection. J Clin Virol 104520. https://doi.org/10.1016/j.jcv.2020.104520
Cepheid Inc. (2020) Xpert® Xpress SARS-CoV-2 [Internet];doi: https://www.cepheid.com/coronavirus
Cepheid Inc. (2020) Xpert® Xpress SARS-CoV-2 Instructions for use [Internet]. doi: https://www.fda.gov/media/136314/download
Pyrc K, Milewska A, Potempa J (2011) Development of loop-mediated isothermal amplification assay for detection of human coronavirus-NL63. J Virol Methods 175:133–136. https://doi.org/10.1016/j.jviromet.2011.04.024
Shirato K, Yano T, Senba S, Akachi S, Kobayashi T, Nishinaka T et al (2014) Detection of Middle East respiratory syndrome coronavirus using reverse transcription loop-mediated isothermal amplification (RT-LAMP). Virol J 11:1–11. https://doi.org/10.1186/1743-422X-11-139
Bhadra S, Jiang YS, Kumar MR, Johnson RF, Hensley LE, Ellington AD (2015) Real-time sequence-validated loop-mediated isothermal amplification assays for detection of Middle East respiratory syndrome coronavirus (MERS-CoV). PLoS One 10:1–21. https://doi.org/10.1371/journal.pone.0123126
Lee SH, Baek YH, Kim YH, Choi YK, Song MS, Ahn JY (2017) One-pot reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) for detecting MERS-CoV. Front Microbiol 7:1–13. https://doi.org/10.3389/fmicb.2016.02166
Huang P, Wang H, Cao Z, Jin H, Chi H, Zhao J et al (2018) A rapid and specific assay for the detection of MERS-CoV. Front Microbiol 9:1–9. https://doi.org/10.3389/fmicb.2018.01101
Shirato K, Semba S, El-Kafrawy SA, Hassan AM, Tolah AM, Takayama I et al (2018) Development of fluorescent reverse transcription loop-mediated isothermal amplification (RT-LAMP) using quenching probes for the detection of the Middle East respiratory syndrome coronavirus. J Virol Methods 258:41–48. https://doi.org/10.1016/j.jviromet.2018.05.006
Kim JH, Kang M, Park E, Chung DR, Kim J, Hwang ES (2019) A simple and multiplex loop-mediated isothermal amplification (LAMP) assay for rapid detection of SARS-CoV. Biochip J 13:341–351. https://doi.org/10.1007/s13206-019-3404-3
Yan C, Cui J, Huang L, Du B, Chen L, Xue G et al (2020) Rapid and visual detection of 2019 novel coronavirus (SARS-CoV-2) by a reverse transcription loop-mediated isothermal amplification assay. Clin Microbiol Infect. https://doi.org/10.1016/j.cmi.2020.04.001
Zhang Y, Odiwuor N, Xiong J, Sun L, Nyaruaba RO, Wei H et al (2020) Rapid molecular detection of SARS-CoV-2 (COVID-19) virus RNA using colorimetric LAMP. medRxiv. https://doi.org/10.1101/2020.02.26.20028373
Zhu X, Wang X, Han L, Chen T, Wang L, Li H, et al. (2020) Reverse transcription loop-mediated isothermal amplification combined with nanoparticles-based biosensor for diagnosis of COVID-19. medRxiv
Butt AM, Siddique S, An X, Tong Y (2020) Development of a dual-gene loop-mediated isothermal amplification (LAMP) detection assay for SARS-CoV-2: a preliminary study. medRxiv. https://doi.org/10.1101/2020.04.08.20056986
Lu R, Wu X, Wan Z, Li Y, Zuo L, Qin J et al (2020) Development of a novel reverse transcription loop-mediated isothermal amplification method for rapid detection of SARS-CoV-2. Virol Sin. https://doi.org/10.1007/s12250-020-00218-1
Broughton JP, Deng W, Fasching CL, Singh J, Charles Y, Chen JS (2020) A protocol for rapid detection of the 2019 novel coronavirus SARS-CoV-2 using CRISPR diagnostics: SARS-CoV-2 DETECTR
El Wahed AA, Patel P, Heidenreich D, Hufert FT, Weidmann M (2013) Reverse transcription recombinase polymerase amplification assay for the detection of middle east respiratory syndrome coronavirus. PLoS Curr ;5. doi: https://doi.org/10.1371/currents.outbreaks.62df1c7c75ffc96cd59034531e2e8364
Koo B, Jin CE, Lee TY, Lee JH, Park MK, Sung H et al (2017) An isothermal, label-free, and rapid one-step RNA amplification/detection assay for diagnosis of respiratory viral infections. Biosens Bioelectron 90:187–194. https://doi.org/10.1016/j.bios.2016.11.051
Koo B, Hong KH, Jin CE, Kim JY, Kim S-H, Shin Y (2018) Arch-shaped multiple-target sensing for rapid diagnosis and identification of emerging infectious pathogens. Biosens Bioelectron 119:79–85. https://doi.org/10.1016/j.bios.2018.08.007
Curti L, Pereyra-Bonnet F, Gimenez C (2020) An ultrasensitive, rapid, and portable coronavirus SARS-CoV-2 sequence detection method based on CRISPR-Cas12. bioRxiv. https://doi.org/10.1101/2020.02.29.971127
Ding X, Yin K, Li Z, Liu C (2020) All-in-one dual CRISPR-Cas12a (AIOD-CRISPR) assay: a case for rapid, ultrasensitive and visual detection of novel coronavirus SARS-CoV-2 and HIV virus. bioRxiv. https://doi.org/10.1101/2020.03.19.998724
Go YY, Kim YS, Cheon S, Nam S, Ku KB, Kim M et al (2017) Evaluation and clinical validation of two field–deployable reverse transcription-insulated isothermal PCR assays for the detection of the Middle East respiratory syndrome–coronavirus. J Mol Diagn 19:817–827. https://doi.org/10.1016/j.jmoldx.2017.06.007
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:1–12. https://doi.org/10.1186/1746-4811-3-12
Freeman WM, Walker SJ, Vrana KE (1999) Quantitative RT-PCR: pitfalls and potential. Biotechniques 26:112–125
Wong ML, Medrano JF (2005) Real-time PCR for mRNA quantitation. Biotechniques 39:75–85
Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582. https://doi.org/10.1038/nprot.2006.236
Deepak SA, Kottapalli KR, Rakwal R, Oros G, Rangappa KS, Iwahashi H et al (2007) Real-time PCR : revolutionizing detection and expression analysis of genes. Curr Genomics 8:234–251
Jothikumar P, Hill V, Narayanan J (2009) Design of FRET-TaqMan probes for multiplex real-time PCR using an internal positive control. Biotechniques 46:519–524
Corman V, Bleicker T, Brünink S, Drosten C, Zambon M (2020) Diagnostic detection of 2019-nCoV by real-time RT-RCR. World Health Organ
Tasrip NA, Khairil Mokhtar NF, Hanapi UK, Abdul Manaf YN, Ali ME, Cheah YK et al (2019) Loop mediated isothermal amplification ; a review on its application and strategy in animal species authentication of meat based food products. Int Food Res J 26:1–10
Becherer L, Borst N, Bakheit M, Frischmann S, Zengerle R, Stetten F Von. Loop-mediated isothermal amplification (LAMP) – review and classification of methods for sequence- specific detection. Anal Methods 2020;doi: https://doi.org/10.1039/c9ay02246e
Zhang X, Lowe SB, Justin J (2014) Brief review of monitoring methods for loop-mediated isothermal ampli fi cation (LAMP). Biosens Bioelectron 61:491–499. https://doi.org/10.1016/j.bios.2014.05.039
Piepenburg O, Williams CH, Stemple DL, Armes NA (2006) DNA detection using recombination proteins. PLoS Biol 4:1115–1121. https://doi.org/10.1371/journal.pbio.0040204
Amer HM, Abd El Wahed A, Shalaby MA, Almajhdi FN, Hufert FT, Weidmann M (2013) A new approach for diagnosis of bovine coronavirus using a reverse transcription recombinase polymerase amplification assay. J Virol Methods 193:337–340. https://doi.org/10.1016/j.jviromet.2013.06.027
Tsai YL, Wang HTT, Chang HFG, Tsai CF, Lin CK, Teng PH, et al. (2012) Development of TaqMan probe-based insulated isothermal PCR (iiPCR) for sensitive and specific on-site pathogen detection. PLoS One ;7. doi: https://doi.org/10.1371/journal.pone.0045278
Liu X, Cheng Z, Fan H, Ai S, Han R (2011) Electrochemical detection of avian influenza virus H5N1 gene sequence using a DNA aptamer immobilized onto a hybrid nanomaterial-modified electrode. Electrochim Acta 56:6266–6270. https://doi.org/10.1016/j.electacta.2011.05.055
Dong S, Zhao R, Zhu J, Lu X, Li Y, Qiu S et al (2015) Electrochemical DNA biosensor based on a tetrahedral nanostructure probe for the detection of avian influenza a (H7N9) virus. ACS Appl Mater Interfaces 7:8834–8842. https://doi.org/10.1021/acsami.5b01438
Picuri JM, Frezza BM, Ghadiri MR (2009) Universal translators for nucleic acid diagnosis. J Am Chem Soc 131:9368–9377. https://doi.org/10.1021/ja902490x
Laschi S, Palchetti I, Marrazza G, Mascini M (2009) Enzyme-amplified electrochemical hybridization assay based on PNA, LNA and DNA probe-modified micro-magnetic beads. Bioelectrochemistry 76:214–220. https://doi.org/10.1016/j.bioelechem.2009.02.012
Nucleic SS, Hybridization A (2000) A theoretical consideration. Basic Tech Mol Biol:221–232. https://doi.org/10.1007/978-3-642-56968-5_10
Abad-Valle P, Fernández-Abedul MT, Costa-García A (2005) Genosensor on gold films with enzymatic electrochemical detection of a SARS virus sequence. Biosens Bioelectron 20:2251–2260. https://doi.org/10.1016/j.bios.2004.10.019
Martínez-Paredes G, González-García MB, Costa-garcía A (2009) Genosensor for SARS virus detection based on gold nanostructured screen-printed carbon electrodes. Electroanalysis 21:379–385. https://doi.org/10.1002/elan.200804399
Díaz-González M, de la Escosura-Muñiz A, González-García MB, Costa-García A (2008) DNA hybridization biosensors using polylysine modified SPCEs. Biosens Bioelectron 23:1340–1346. https://doi.org/10.1016/j.bios.2007.12.001
González-lópez A, Abedul MTF (2020) Genosensor on gold films with enzymatic electrochemical detection of a SARS virus sequence [Internet]. Elsevier Inc. doi: https://doi.org/10.1016/B978-0-12-815932-3.00021-8
Jung IY, You JB, Choi BR, Kim JS, Lee HK, Jang B et al (2016) A highly sensitive molecular detection platform for robust and facile diagnosis of Middle East respiratory syndrome (MERS) Corona virus. Adv Healthc Mater 5:2168–2173. https://doi.org/10.1002/adhm.201600334
Kim H, Park M, Hwang J, Kim JH, Chung D-R, Lee K-S et al (2019) Development of label-free colorimetric assay for MERS-CoV using gold nanoparticles. ACS Sensors 4:1306–1312. https://doi.org/10.1021/acssensors.9b00175
Teengam P, Siangproh W, Tuantranont A, Vilaivan T, Chailapakul O, Henry CS (2017) Multiplex paper-based colorimetric DNA sensor using pyrrolidinyl peptide nucleic acid-induced AgNPs aggregation for detecting MERS-CoV, MTB and HPV oligonucleotides. Anal Chem 86:5428–5435. https://doi.org/10.1021/acs.analchem.7b00255
Li H, Rothberg L, Austin RH (2004) Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc Natl Acad Sci 101:14036–14039. https://doi.org/10.1073/pnas.0406115101
Yang JM, Kim KR, Kim CS (2018) Biosensor for rapid and sensitive detection of influenza virus. Biotechnol Bioprocess Eng 23:371–382. https://doi.org/10.1007/s12257-018-0220-x
Hassanpour S, Baradaran B, Hejazi M, Hasanzadeh M, Mokhtarzadeh A, de la Guardia M (2018) Recent trends in rapid detection of influenza infections by bio and nanobiosensor. TrAC Trends Anal Chem 98:201–215. https://doi.org/10.1016/j.trac.2017.11.012
Caygill RL, Blair GE, Millner PA (2010) A review on viral biosensors to detect human pathogens. Anal Chim Acta 681:8–15. https://doi.org/10.1016/j.aca.2010.09.038
Ilkhani H, Farhad S (2018) A novel electrochemical DNA biosensor for Ebola virus detection. Anal Biochem 557:151–155. https://doi.org/10.1016/j.ab.2018.06.010
Damborský P, Švitel J, Katrlík J (2016) Optical biosensors. Essays Biochem 60:91–100. https://doi.org/10.1042/EBC20150010
Vashist SK (2020) In vitro diagnostic assays for COVID-19: recent advances and emerging trends. Diagnostics 10:202. https://doi.org/10.3390/diagnostics10040202
Sun C, Chen L, Yang J, Luo C, Zhang Y, Li J et al (2020) SARS-CoV-2 and SARS-CoV Spike-RBD structure and receptor binding comparison and potential implications on neutralizing antibody and vaccine development. bioRxrv. https://doi.org/10.1101/2020.02.16.951723
Li X, Geng M, Peng Y, Meng L, Lu S (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. https://doi.org/10.1016/j.jpha.2020.03.001
Grant PR, Garson JA, Tedder RS, Chan PK, Tam JS, Sung JJ (2003) Detection of SARS coronavirus in plasma by real-time RT-PCR. N Engl J Med 349:2468–2469. https://doi.org/10.1056/NEJM200312183492522
Paules CI, Marston HD, Fauci AS (2020) Coronavirus infections-more than just the common cold. JAMA ;323:707–8. doi: 10.1007/82
Lau SKP, Woo PCY, Wong BHL, Tsoi H-W, Woo GKS, Poon RWS et al (2004) Detection of severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein in SARS patients by enzyme-linked immunosorbent assay. J Clin Microbiol 42:2884–2889. https://doi.org/10.1128/JCM.42.7.2884
Zhai J, Briese T, Dai E, Wang X, Pang X, Du Z et al (2004) Real-time polymerase chain reaction for detecting SARS coronavirus, Beijing, 2003. Emerg Infect Dis 10:311. https://doi.org/10.3201/eid1002.030799
Sunwoo HH, Palaniyappan A, Ganguly A, Bhatnagar PK, Das D, El-Kadi AOS et al (2013) Quantitative and sensitive detection of the SARS-CoV spike protein using bispecific monoclonal antibody-based enzyme-linked immunoassay. J Virol Methods 187:72–78. https://doi.org/10.1016/j.jviromet.2012.09.006
Layqah LA, Eissa S (2019) An electrochemical immunosensor for the corona virus associated with the Middle East respiratory syndrome using an array of gold nanoparticle-modified carbon electrodes. Microchim Acta 186:224. https://doi.org/10.1007/s00604-019-3345-5
Mahari S, Roberts A, Shahdeo D, Gandhi S (2020) eCovSens-ultrasensitive novel in-house built printed circuit board based electrochemical device for rapid detection of nCovid-19. bioRxiv. https://doi.org/10.1101/2020.04.24.059204
Zhang X, Qi Q, Jing Q, Ao S, Zhang Z, Ding M, et al. (2020) Electrical probing of COVID-19 spike protein receptor binding domain via a graphene field-effect transistor. arXiv:2003.12529 ;1–20
Zuo B, Li S, Guo Z, Zhang J, Chen C (2004) Piezoelectric Immunosensor for SARS-associated coronavirus in sputum. Anal Chem 76:3536–3540. https://doi.org/10.1021/ac035367b
Chang Y-F, Huang JC, Su L-C, Chen Y-MA, Chen C-C, Chou C (2009) Localized surface plasmon coupled fluorescence fiber-optic biosensor for severe acute respiratory syndrome coronavirus nucleocapsid protein detection. 2009 14th Optoelectron. Commun. Conf. 1–2, IEEE doi: https://doi.org/10.1109/OECC.2009.5215723
Diao B, Wen K, Chen J, Liu Y, Yuan Z, Han C et al (2020) Diagnosis of acute respiratory syndrome coronavirus 2 infection by detection of nucleocapsid protein. medRxiv. https://doi.org/10.1101/2020.03.07.20032524
Huang JC, Chang Y, Chen K, Su L, Lee C, Chen C et al (2009) Detection of severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein in human serum using a localized surface plasmon coupled fluorescence fiber-optic biosensor. Biosens Bioelectron 25:320–325. https://doi.org/10.1016/j.bios.2009.07.012
Chen Y, Chan K-H, Kang Y, Chen H, Luk HKH, Poon RWS et al (2015) A sensitive and specific antigen detection assay for Middle East respiratory syndrome coronavirus. Emerg Microbes Infect 4:e26. https://doi.org/10.1038/emi.2015.26
Kammila S, Das D, Bhatnagar PK, Sunwoo HH, Zayas-Zamora G, King M et al (2008) A rapid point of care immunoswab assay for SARS-CoV detection. J Virol Methods 152:77–84. https://doi.org/10.1016/j.jviromet.2008.05.023
Chen Y, Chan K-H, Hong C, Kang Y, Ge S, Chen H et al (2016) A highly specific rapid antigen detection assay for on-site diagnosis of MERS. J Inf Secur 73:82–77. https://doi.org/10.1016/j.jinf.2016.04.014
Fung J, Lau SKP, Woo PCY (2020) Antigen capture enzyme-linked immunosorbent assay for detecting Middle East respiratory syndrome coronavirus in humans. In: MERS Coronavirus. Humana, New York, pp 89–97. https://doi.org/10.1007/978-1-0716-0211-9_7
Che X-Y, Qiu L-W, Liao Z-Y, Wang Y-D, Wen K, Pan Y-X et al (2005) Antigenic cross-reactivity between severe acute respiratory syndrome-associated coronavirus and human coronaviruses 229E and OC43. J Infect Dis 191:2033–2037. https://doi.org/10.1086/430355
Quidel Corp (2020) Sofia SARS antigen FIA for in vitro diagnostic use [Internet]. doi: https://www.fda.gov/media/137885/download
Mettler K (2020) FDA issues emergency approval of new antigen test that is cheaper, faster and simpler [Internet]. Washington Post ;doi: https://www.washingtonpost.com/health/2020/05/09/fda-issues-emergency-approval-new-antigen-test-that-is-cheaper-faster-simpler/
Ishikawa FN, Chang H-K, Curreli M, Liao H-I, Olson CA, Chen P-C et al (2009) Label-free, electrical detection of the SARS virus N-protein with nanowire capture probes. ACS Nano 3:1219–1224. https://doi.org/10.1021/nn900086c
Ishikawa FN, Curreli M, Olson CA, Liao H-I, Sun R, Roberts RW et al (2010) Importance of controlling nanotube density for highly sensitive and reliable biosensors functional in physiological conditions. ACS Nano 4:6914–6922. https://doi.org/10.1021/nn101198u
Yang Y, Wang Q, Guo D (2008) A novel strategy for analyzing RNA-protein interactions by surface plasmon resonance biosensor. Mol Biotechnol 40:87–93. https://doi.org/10.1007/s12033-008-9066-3
Ahn D-G, Jeon I-J, Kim JD, Song M-S, Han S-R, Lee S-W et al (2009) RNA aptamer-based sensitive detection of SARS coronavirus nucleocapsid protein. Analyst 134:1896–1901. https://doi.org/10.1039/b906788d
Roh C, Jo SK (2011) Quantitative and sensitive detection of SARS coronavirus nucleocapsid protein using quantum dots-conjugated RNA aptamer on chip. J Chem Technol Biotechnol 86:1475–1479. https://doi.org/10.1002/jctb.2721
Li M, Jin R, Peng Y, Wang C, Ren W, Lv F et al (2020) Generation of antibodies against COVID-19 virus for development of diagnostic tools. medRxiv. https://doi.org/10.1101/2020.02.20.20025999
Ibau C, Arshad MM, Gopinath SCB (2017) Current advances and future visions on bioelectronic immunosensing for prostate-specific antigen. Biosens Bioelectron 98:267–284. https://doi.org/10.1016/j.bios.2017.06.049
Afzal A, Mujahid A, Schirhagl R, Bajwa SZ, Latif U, Feroz S (2017) Gravimetric viral diagnostics: QCM based biosensors for early detection of viruses. Chemosensors 5:7. https://doi.org/10.3390/chemosensors5010007
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shabani, E., Dowlatshahi, S. & Abdekhodaie, M.J. Laboratory detection methods for the human coronaviruses. Eur J Clin Microbiol Infect Dis 40, 225–246 (2021). https://doi.org/10.1007/s10096-020-04001-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-020-04001-8