
Abstract
The purpose of this study was to describe epidemiological, clinical and microbiological characteristics of confirmed novel influenza A (H1N1) infection, investigating factors associated with disease severity. We retrospectively selected patients seeking care for respiratory symptoms in two periods (May–August and September–November 2009) with different epidemiological characteristics. Only patients with confirmed pandemic influenza A (H1N1) were enrolled in this study. A total of 104 patients with H1N1 infection were evaluated, mostly referring classic influenza symptoms; in addition, diarrhea and vomiting were often referred. Clinical signs, symptoms and respiratory complications were different in the two periods. Of all patients, 18 (17%) had pneumonia. Patients older than 50 years showed a lower probability of pneumonia diagnosis when compared to children aged 0–13 (p = 0.049); a longer duration of symptoms before medical care was associated with a higher probability of pneumonia (p = 0.026). Phylogenetic analysis showed a low variability both in hemagglutinin and neuraminidase genes. In addition, no neuraminidase mutation associated with antiviral resistance was detected. A detailed description of respiratory diseases associated with H1N1 infection was provided and factors associated with its severity were investigated, thus contributing to the insight into epidemiological, clinical and microbiological knowledge of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza A H1N1 virus is a subtype of influenza A virus, the most common cause of influenza (flu) in humans. Some strains of H1N1 are endemic in humans and cause a small fraction of all seasonal influenza or influenza-like illnesses. H1N1 strains caused a tiny percent of all human flu infections in 2004–2005 but other H1N1 strains are endemic in pigs (swine influenza) and in birds (avian influenza) [1].
The influenza pandemic the world was waiting for might have arrived on April 2009. Mexico was the first country where a sharp increase in reports of patients with pneumonia caused by influenza A H1N1 was observed [2]. In June 2009, the World Health Organization (WHO) declared that flu due to a new strain of swine-origin H1N1 was responsible for the 2009 influenza pandemic. According to the Centers for Disease Control and Prevention (CDC), in humans the symptoms of the 2009 "swine flu" H1N1 virus were similar to those of seasonal influenza and influenza-like illness in general, including fever, cough, sore throat, headache, chills and fatigue. Preliminary reports have shown that, when compared to previous influenza seasons, the 2009 outbreak showed an increased percentage of patients reporting diarrhea and vomiting [3]. The most common cause of death was respiratory failure. Other reported causes of death included pneumonia, high fever (leading to neurological problems), dehydration (from excessive vomiting and diarrhea) and electrolyte imbalance. Severe cases were most frequent in middle-aged patients, often with comorbidities [4]. However, to date the virulence of H1N1 strains appears to be no greater than that of seasonal influenza.
The aim of this study was to describe epidemiological, clinical and microbiological characteristics of all confirmed novel influenza A H1N1 cases observed in a reference hospital in Rome from May to November 2009, focusing on the impact that seasonal and epidemiological factors could have on clinical features of the disease.
Materials and methods
Patients
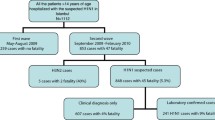
Patients admitted with respiratory symptoms and/or fever (temperature >38.5°C) at the Emergency Room of the Catholic University of Sacred Heart in Rome, in two different periods of the year (May–August and September–November 2009) were retrospectively selected. The separation of the cases in the two periods was justified by epidemiological factors. During the first period H1N1 pandemic virus was not epidemic in the country and the Italian Ministry of Health adopted a policy aimed at containing its spread from travellers and their contacts, recommending testing for the new influenza virus in all probable cases of influenza A H1N1 (defined as those showing typical symptoms and having had recent contact with proven cases of H1N1, or returning from travel to areas where influenza A H1N1 was epidemic or working in a laboratory) observed in reference hospitals. In the second period, when the virus was endemic in Italy, the definition of probable cases was less stringent, without the inclusion of epidemiological criteria; probable cases included patients with fever greater than or equal to 38°C associated with at least one constitutional symptom (headache, asthenia, tremors, weakness, abdominal pain and diarrhea) and accompanied by at least one respiratory symptom (cough, sore throat, nasal congestion). However, in this period H1N1 testing was recommended only for subjects with more severe disease or requiring hospitalization.
Only patients with pandemic influenza A (H1N1) virus detected from nasal or oropharingeal swabs by real-time RT PCR assay were considered for this analysis.
Real-time RT PCR and viral sequencing
A 400-μl aliquot of the specimen was used for automated RNA extraction with a EZ1 viral kit (QIAGEN, Hilden, Germany) for use in real-time RT PCR. The primers and probes for the H1 gene (swH1) and M gene (InfA) used in this work were recommended by WHO [5] and synthesized by the Applied Biosystems (Forest City, CA, USA) company. RT-PCR was performed in a 25-μl reaction volume that contained 5 μl of the RNA dilution, 12.5-μl 2x AgPath-ID™ One-Step RT-PCR buffer, 1 μl enzyme mix, and 0.5 μl assay mix in a fluorometric PCR instrument (ABI 7300). Thermal cycling conditions were 30 min at 50°C followed by 10 min at 95°C and a subsequent 45 cycles amplification (95°C for 15 s; 55°C for 30 s, fluorescence was collected at 55°C).
In all patients with H1N1 infection confirmed by RT-PCR on respiratory specimens, samples stored at −80°C were then grown in MDCK cell cultures for viral isolation and subsequent HA and NA genes sequencing. Amplicons for sequencing were generated by reverse transcription, followed by PCR amplification to generate overlapping double-stranded DNA amplicons covering HA and NA segments of the influenza virus genome [3], the products were purified with QIAquick columns (Qiagen) and sequenced by using an ABI Prism BigDye Terminator (version 1.1) cycle sequencing kit in an ABI Prism 3100-genetic analyzer on a 50-cm array. The sequencing reaction was performed according to the manufacturer's protocol in a final volume of 10 μl with approximately 2 ng of amplified product, BigDye Terminator (version 1.1) premix, and 0.3 pmol of the primer. Unincorporated nucleotides were purified by using a BigDye XTerminator purification kit (ABI). The data were collected by using ABI software (version 2.0) [6]. The sequences were analyzed by using Seqman software (Lasergene package).
Phylogenetic analysis
Sequenced HA and NA genes were used for a BLAST search in the NCBI Genbank and an additional set of background sequences with high similarity was collected, adding a reference for H1N1 and an outgroup of H2N2 strain. Sequences were aligned using Opal software [7]. Maximum likelihood (ML) phylogenetic analysis was performed using parallel computational methods, setting up Akaike Information Criterion (AIC)-based model selection, nearest-neighbor-interchange tree search, four gamma categories, and 100 bootstrap [8]. Sequence variability was annotated using entropy plots.
Statistical analysis
Characteristics of the patients who presented in the two periods May–August and September–November were compared using Fisher’s exact test or chi-square test for categorical variables and the Student’s t test for continuous variables. Denominators that were used to calculate proportions varied according to the number of patients with available data. Factors associated with pneumonia diagnosis were analyzed by univariable and multivariable logistic regression. A two-sided P value of less than 0.05 was considered statistically significant. All analyses were performed with SPSS version 18.0 (SPSS Inc, Chicago, IL).
Results
Patient characteristics
A total of 104 patients (52% males, median age 15 years [interquartile range, IQR 13–25]) with H1N1 infection were evaluated. Of these, 77 (74%) were admitted to the Emergency ward during May–August 2009 and 27 (26%) during September–November 2009. Overall, 42 (40%) patients reported travelling in European countries (outside Italy), 14 (14%) in the United States and six (6%) in other geographical areas, while 42 (40%) had never left Italy. As expected, in the second period a significant increase in the number of patients not reporting travelling outside Italy was observed (96% versus 19%, p < 0.001). Patient characteristics according to the subgroups are summarized in Table 1.
Clinical presentation: symptoms, signs and laboratory parameters
A description of symptoms, signs and laboratory parameters according to subgroups is summarized in Table 2.
In our series, fever was reported by 102 (98%) patients, dry cough by 52 (50%) and productive cough by 17 (16%), vomiting by six (6%) and diarrhea by five (5%). Other symptoms reported by patients included sore throat (34%), asthenia (11%), headache (11%), arthralgia (8%), myalgia (7%), conjunctivitis (6%), abdominal pain (4%), wheezing (4%) and cyanosis (2%). A total of 17 patients (16%) required hospitalization, two had severe respiratory failure and required admission to the intensive care unit. In particular, among hospitalized patients, four (23%) were subjects under the age of 14 years, seven (42%) between 15 and 39 years and six (35%) older than 40 years. When hospitalization rate was analyzed for each age group, we found that it was higher in patients older than 40 years when compared to those aged 15–39 (13% versus 46%, p = 0.008) or to those under 14 years (11% versus 46%, p = 0.006). None of the patients died. Oseltamivir (at a dosage of 75 mg bid for 5 days) was administered within 48 hours after the onset of symptoms in 22 (22%) patients. Thirteen (12%) subjects had received outpatient antimicrobial therapy while 20 (20%) were prescribed antibiotics after medical care in the emergency room. Three (3%) patients had pre-existing predisposing disorders (one patient had asthma and heart disease, two patients immunosuppression.
Blood tests were performed in 89 (86%) individuals. Of these, nine (10%) patients had leukocytosis, seven (8%) had leukopenia, 36 (40%) had lymphopenia and four (4%) had lymphocytosis.
Clinical signs and symptoms on admission and the respiratory complications significantly differed from the first to the second period with dry cough declining from 56% to 33% (p = 0.04), sore throat declining from 42% to 11% (p = 0.004), and vomiting increasing from 3% to 15% (p = 0.01). Pneumonia diagnosis was more frequent in the second period (37% versus 10%, p = 0.002). Hospitalization rate increased from 8% to 41% (p < 0.001) and admission to intensive care, which was absent in the first period, occurred in 11% of the cases observed during the second period (p = 0.003). Prescription of antibiotic therapy increased from 8% to 64% (p < 0.001) and use of oseltamivir from 13% to 54% (p < 0.001).
Clinical complications
In total, 18 cases (17%) had pneumonia confirmed by chest X-ray. Twelve cases (67%) showed focal infiltrates involving a single lung and six (33%) showed a bilateral interstitial-alveolar pattern. In all these patients blood cultures and culture of respiratory specimens were negative. Among patients with pneumonia, four (27%) had leukocytosis (leukocyte count >9,800 per cubic millimeter) and three (20%) had lymphopenia (absolute lymphocyte count <1,680 per cubic millimeter). Five (28%) patients started early antimicrobial therapy before access to the Emergency Department. Overall, antibiotics were administered to nine (50%) subjects. Oral levofloxacin and amoxicillin-clavulanic acid were the most frequently prescribed drugs.
At univariable analysis, older age, a negative epidemiology for recent foreign travel, emergency admission between September and November 2009, longer time from onset symptoms to emergency admission, treatment with antibiotic therapy and oseltamivir were all associated with higher odds of pneumonia. No specific symptoms, signs or laboratory parameters were associated with the occurrence of pneumonia. Due to the small sample size, a full multivariable analysis (including all variables that showed an association with pneumonia at univariate analysis) could not be performed since it would have exhibited a low statistical power. We thus executed single sets of bivariable logistic regression analyses, including only variables considered clinically relevant and excluding variables such as the use of antimicrobial therapy and oseltamivir administration since these were constant in the clinical management of pneumonia and rather a consequence than a cause of pneumonia (see Table 3). At multivariate analysis, patients older than 50 years showed a lower probability of pneumonia diagnosis when compared to children aged 0–13 (p = 0.049); a longer duration of symptoms before medical care was associated with a higher probability of pneumonia (p = 0.026).
Sequence and phylogenetic analysis
Overall, 63 and 46 sequences could be obtained for phylogenetic analysis of HA and NA genes, respectively. By a BLAST search on the Genbank database, keeping the first best-scoring sequences for each one of our sequences and eliminating duplicates, an additional set of 12 and 15 sequences was added to HA and NA sets. The reference H1 and N1 sequences (CY033577 and CY033579) from Puerto Rico were added, as well as the outgroup references H2 and N2 (NC_007374 and NC_007382) from Korea. Five NA sequences with reported resistance to oseltamivir were eventually added to the NA set. In the HA gene, BLAST search reported that the best-scoring sequences were distributed in several countries all over the world (Russia, Turkey, Germany, China, Canada, Sweden, Taiwan, Netherlands, Thailand). The same was when considering the NA gene (Argentina, California, Canada, Italy, Russia, Malaysia, United States) and two sequences from Italy (Ancona province). After multiple alignment and gap-stripping, entropy analysis on the nucleotide alignment showed very low variation (Fig. 1a, b): the average (stdev) entropy value for HA was 0.005 (0.04) and the median (IQR) was 0 (0–0); the average (stdev) entropy value for NA was 0.004 (0.03) and the median (IQR) was 0 (0–0). The analysis of the deduced aminoacid sequences revealed no changes associated with oseltamivir or zanamivir resistance (E119V, H274Y, R292K, N294S) in the NA gene. The bootstrapped ML phylogenetic analysis, highly affected by the extreme sequence identity, revealed no evidence of clustering in the HA gene (selected model via AIC was general time reversible), and the same held in the NA gene (selected model via AIC was transversional model). Figure 2a, b depicts the relative phylogenetic trees.

a Entropy plot on gap-stripped nucleotide alignment for hemagglutinin (HA) gene. b Entropy plot on gap-stripped nucleotide alignment for neuraminidase (NA) gene

Phylogenetic trees for hemagglutinin (HA, n = 63) gene (a) and neuraminidase (NA, n = 46) gene (b). Trees were estimated via maximum-likelihood, and 100 bootstrap replicates. An additional set of background sequences with high similarity was added (by a BLAST search on genbank, n = 12 to the HA set and n = 15 to the NA set), along with two reference H1 and N1 sequences (CY033577 and CY033579), five NA sequences with resistance to oseltamivir, plus an outgroup of H2N2 strain
Discussion
The global and rapid spread of pandemic influenza A H1N1 raised concerns for the serious complications potentially associated with infection. Reports from different countries describing clinical features of H1N1 infection have been published [9–11]; however, several aspects (such as differences in epidemiological trends and in clinical presentation between different countries or different seasons) are still unexplored. As a consequence, further surveillance is needed.
In this study, patients with pandemic 2009 H1N1 virus infection diagnosed in Italy during two periods (May–August and September–November 2009) characterized by different epidemiological features were enrolled. Regarding clinical characteristics, influenza A H1N1 infected patients mostly referred the classic influenza symptoms; in addition, H1N1 patients often had diarrhea and vomiting, which are not usually seen in seasonal influenza [3]. In our series, hospitalization was required by 16% of patients, the majority of which were aged less than 40 years, confirming that children and young adults were the age category mostly affected by the epidemic [12], probably resulting from the higher susceptibility of the youngest to the new virus due to the lack of H1N1-specific antibodies or memory immune response [13]. This is in contrast with epidemiological features of seasonal influenza, for which hospitalizations are more common among persons 65 years of age or older and those under the age of 15 years. However, hospitalization rate was higher in patients older than 40 years, suggesting that when H1N1 disease occurs in this age group it can be more severe.
Risk factors for acquiring the infection or its complications significantly varied between the two explored periods. In particular, a history of travel in a country where endemic transmission of H1N1 virus occurred was most frequently reported in the first period, while pre-existing conditions predisposing to clinical severity were prevalent in patients observed in the second period. Moreover, several complications, such as pneumonia, wheezing, hospitalization or admission to an intensive care unit, were more frequently observed in the second period. These observations may also reflect the change in health policies when H1N1 strains became endemic in Italy, with authorities recommending influenza testing only for patients with more severe respiratory disease. However, the influence of several other factors (such as the role of cold climate in favoring complications, a higher diffusion of the virus in age groups with risk factors for complications or a increased virulence of circulating strains) cannot be fully excluded.
Few studies analyzed factors predisposing to the development of clinical complications and further investigation is required. In our series, we observed that children and those with a longer duration of symptoms before medical care showed a higher risk of pneumonia and, as a consequence, should be carefully evaluated for the possible development of such complication.
Phylogenetic analysis is a useful tool to investigate whether differences in clinical presentation between different seasons or different countries could be attributable to viral evolution and to define and understand dynamics of the evolution of viral epidemics. In our study population, sequence analysis showed that the nucleotide and amino acidic variability was low both in the HA and the NA genes. In addition, we did not detect any virus showing mutations in the NA gene that are associated with antiviral resistance. This is not unexpected, given the generally low antiviral uptake in Italy and Europe. During the 2008–2009 seasonal influenza, there was a dramatic increase in the oseltamivir-resistant strain circulation in some countries [14]. In addition, phylogenetic analysis did not show any clear clustering, although a differentiation from the original Puerto Rico strain occurred. The BLAST search on the Genbank database reported that the sequences showing the best associations with those observed in this study were widely distributed in the world, in agreement with the epidemiological information for which many of the diagnosed cases were subjects who travelled outside the country. This may suggest that the epidemic in the country was probably the result of multiple entry points. Notwithstanding, it must be considered that the evidence of no-clustering suggests that the evolution of H1N1 probably acts at a higher epidemic level and across the whole viral genome. In fact, a recent study showed that H1N1 is evolving into two distinct clusters [15]. Another paper, investigating viral phylogeography, highlighted that the H1N1 drift needs to be analyzed with respect to the whole genome [16] in a worldwide or continental setting. A limitation of this study is that only a portion of the H1N1 genome was sequenced (HA and NA genes) and it was not possible to obtain both the HA and NA genes for all the patients. Due to the low genetic variability observed, a full-genome phylogenetic analysis (after ascertaining that the genes are concatenable) of the study population, adding an additional background population representative of the worldwide epidemic, could be advisable.
In conclusion, a detailed description of respiratory diseases associated with H1N1infection was provided and factors associated with its severity were investigated, thus contributing to the insight into epidemiological, clinical and microbiological knowledge of the disease. Further studies are needed to fully understand the evolution of epidemiological features and viral dynamics of H1N1 infection in humans, as well as to provide a basis for planning resources for the development of new vaccine strategies and antiviral agents use.
References
Zimmer SM, Burke DS (2009) Historical perspective—Emergence of influenza A (H1N1) viruses. N Engl J Med 361:279–285
Laguna-Torres VA, Benavides JG (2009) Infection and death from influenza A H1N1 virus in Mexico. Lancet 374:2032–2033
Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ et al (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360:2605–2615
Estenssoro E, Ríos FG, Apezteguía C, Reina R, Neira J, Ceraso DH et al (2010) Pandemic 2009 Influenza A (H1N1) in Argentina: a study of 337 patients on mechanical ventilation. Am J Respir Crit Care Med 182:41–48
World Health Organization (2009) CDC protocol of real-time RT-PCR for influenza A (H1N1). Available at: http://www.who.int/csr/resources/publications/swineflu/CDCRealtimeRTPCR_SwineH1Assay-2009_20090430.pdf. Accessed 12 November 2010
Pabbaraju K, Wong S, Wong AA, Appleyard GD, Chui L, Pang XL et al (2009) Design and validation of real-time reverse transcription-PCR assays for detection of pandemic (H1N1) 2009 virus. J Clin Microbiol 47:3454–3460
Wheeler T, Kececioglu J (2007) Multiple alignment by aligning alignments. Proceedings of the 15th ISCB Conference on Intelligent Systems for Molecular Biology. Bioinformatics 23:i559–i568
Keane TM, Naughton TJ, McInerney JO (2007) A high-throughput phylogenomics webserver using distributed computing. Nucleic Acids Research 35:W33–W37
Jain S, Kamimoto L, Bramley AM, Schmitz AM, Benoit SR, Louie J et al (2009) Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 361:1935–1944
Dominguez-Cherit G, Lapinsky SE, Macias AE, Pinto R, Espinosa-Perez L, De la Torre A et al (2009) Critically ill patients with 2009 influenza A (H1N1) in Mexico. JAMA 302:1880–1887
Cao B, Li XW, Mao Y, Wang J, Lu HZ, Chen YS et al (2009) Clinical features of the initial cases of 2009 pandemic influenza A (H1N1) virus infection in China. N Engl J Med 361:2507–2517
Chowell G, Bertozzi SM, Colchero MA, Lopez-Gatell H, Alpuche-Aranda C, Hernandez M et al (2009) Severe respiratory diseases concurrent with the circulation of H1N1 influenza. N Engl J Med 361:674–679
Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H et al (2009) Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 361:1945–1952
Baranovich T, Saito R, Suzuki Y, Zaraket H, Dapat C, Caperig-Dapat I et al (2010) Emergence of H274Y oseltamivir-resistant A(H1N1) influenza viruses in Japan during the 2008–2009 season. J Clin Virol 47:23–28
Fereidouni SR, Beer M, Vahlenkamp T, Starick E (2009) Differentiation of two distinct clusters among currently circulating influenza A(H1N1)v viruses, March–September 2009. Euro Surveill 14:46
Parks D, Macdonald N, Beiko R (2009) Tracking the evolution and geographic spread of Influenza. A PLoS Curr Influenza 27:RRN1014
Acknowledgements
No specific funding was received for this study.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Di Giambenedetto, S., Zileri Dal Verme, L., Sali, M. et al. Clinical presentation, microbiological features and correlates of disease severity of 2009 pandemic influenza A (H1N1) infection. Eur J Clin Microbiol Infect Dis 30, 541–549 (2011). https://doi.org/10.1007/s10096-010-1116-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-010-1116-7