
Abstract
The mutations of MYH7 (slow skeletal/β-cardiac myosin heavy chain) are commonly found in familial hypertrophic/dilated cardiomyopathy, and also can cause Laing early-onset distal myopathy (LDM), myosin storage myopathy (MSM), and congenital myopathy with fiber-type disproportion (CFTD). Here we report two cases whose diagnosis was hereditary myopathy according to clinical feature and muscle pathology analysis. High-throughput genomic sequencing (next generation sequencing) was performed to validate the diagnosis. Two MYH7 mutations, p.R1845W and p.E1687del, were identified. p.R1845W was found in a male patient showing weakness of both terminal lower legs without foot drop. Muscle pathology stainings characteristically showed the hyaline body in the intracytoplasmic location. The novel mutation p.E1687del was found in a family with seven patients. The proband showed foot drop, scoliosis, and winged scapula, while his mother only showed mild foot drop and winged scapula. Muscle pathology analysis showed congenital centronucleus myopathy. Both cases only showed muscular disorder and had no cardiomyopathy. This study, for the first time, reports the MYH7 mutations associated with centronucleus myopathy.



Similar content being viewed by others

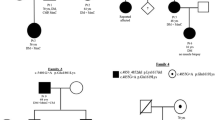
References
Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP (1990) The complete sequence of the human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics 8(2):194–206. https://doi.org/10.1016/0888-7543(90)90272-V
Walsh R, Rutland C, Thomas R, Loughna S (2010) Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115(1):49–60. https://doi.org/10.1159/000252808
Yüceyar N, Ayhan Ö, Karasoy H, Tolun A (2015) Homozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromuscul Disord 25(4):340–344. https://doi.org/10.1016/j.nmd.2015.01.007
Naddaf E and Waclawik, Andrew J. (2015) Two Families With MYH7 Distal Myopathy Associated With Cardiomyopathy and Core Formations. J Clin Neuromusc Dis 16(3):164–169 https://doi.org/10.1097/CND.0000000000000069
Muelas N, Hackman P, Luque H, Garces-Sanchez M, Azorin I, Suominen T, Sevilla T, Mayordomo F, Gomez L, Marti P, Maria Millan J, Udd B, Vilchez JJ (2010) MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology 75(8):732–741. https://doi.org/10.1212/WNL.0b013e3181eee4d5
Overeem S, Schelhaas HJ, Blijham PJ, Grootscholten MI, ter Laak HJ, Timmermans J, van den Wijngaard A, Zwarts MJ (2007) Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord 17(6):490–493. https://doi.org/10.1016/j.nmd.2007.02.007
Laing NG, Ceuterick-de Groote C, Dye DE, Liyanage K, Duff RM, Dubois B, Robberecht W, Sciot R, Martin JJ, Goebel HH (2005) Myosin storage myopathy: slow skeletal myosin (MYH7) mutation in two isolated cases. Neurology 64(3):527–529. https://doi.org/10.1212/01.WNL.0000150581.37514.30
Shingde MV, Spring PJ, Maxwell A, Wills EJ, Harper CG, Dye DE, Laing NG, North KN (2006) Myosin storage (hyaline body) myopathy: a case report. Neuromuscul Disord 16(12):882–886. https://doi.org/10.1016/j.nmd.2006.09.001
Jie L et al (2015) Mutation and clinical relevance in a large cohort of unrelated Chinese patients with hypertrophic cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 43(8):682–689
Liu W, Liu W, Hu D, Zhu T, Ma Z, Yang J, Xie W, Li C, Li L, Yang J, Li T, Bian H, Tong Q (2013) Mutation spectrum in a large cohort of unrelated Chinese patients with hypertrophic cardiomyopathy. Am J Cardiol 112(4):585–589. https://doi.org/10.1016/j.amjcard.2013.04.021
Park JM, Kim YJ, Yoo JH, Hong YB, Park JH, Koo H, Chung KW, Choi BO (2013) A novel MYH7 mutation with prominent paraspinal and proximal muscle involvement. Neuromuscul Disord 23(7):580–586. https://doi.org/10.1016/j.nmd.2013.04.003
Oda T, Xiong H, Kobayashi K, Wang S, Satake W, Jiao H, Yang Y, Cha PC, Hayashi YK, Nishino I, Suzuki Y, Sugano S, Wu X, Toda T (2015) A de novo mutation of the MYH7 gene in a large Chinese family with autosomal dominant myopathy. Human Genome Variation 2:15022. https://doi.org/10.1038/hgv.2015.22
Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C (2007) MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord 17(4):321–329. https://doi.org/10.1016/j.nmd.2007.01.010
Fiorillo C et al (2016) MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis 11(1):91. https://doi.org/10.1186/s13023-016-0476-1
Clarke NF, Amburgey K, Teener J, Camelo-Piragua S, Kesari A, Punetha J, Waddell LB, Davis M, Laing NG, Monnier N, North KN, Hoffman EP, Dowling JJ (2013) A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies. Neuromuscul Disord 23(5):432–436. https://doi.org/10.1016/j.nmd.2013.02.009
Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, Tan SV, Sheehan J, Muntoni F, Abbs S, Sewry CA, Dubowitz V, Laing NG, Jungbluth H (2012) Mutations in MYH7 cause multi-minicore disease (MmD) with variable cardiac involvement. Neuromuscul Disord 22(12):1096–1104. https://doi.org/10.1016/j.nmd.2012.06.007
Olive M (2009) Extralysosomal protein degradation in myofibrillar myopathies. Brain Pathol 19(3):507–515. https://doi.org/10.1111/j.1750-3639.2009.00288.x
Ortolano S, Tarrío R, Blanco-Arias P, Teijeira S, Rodríguez-Trelles F, García-Murias M, Delague V, Lévy N, Fernández JM, Quintáns B, Millán BS, Carracedo Á, Navarro C, Sobrido MJ (2011) A novel MYH7 mutation links congenital fiber type disproportion and myosin storage myopathy. Neuromuscul Disord 21(4):254–262. https://doi.org/10.1016/j.nmd.2010.12.011
Lin P, Liu X, Zhao D, Dai T, Wu H, Gong Y, Yan C (2016) DNM2 mutations in Chinese Han patients with centronuclear myopathy. Neurol Sci 37(6):995–998. https://doi.org/10.1007/s10072-016-2513-1
Chen T, Pu C, Wang Q, Liu J, Mao Y, Shi Q (2015) Clinical, pathological, and genetic features of dynamin-2-related centronuclear myopathy in China. Neurol Sci 36(5):735–741. https://doi.org/10.1007/s10072-014-2028-6
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 17(5):405–423. https://doi.org/10.1038/gim.2015.30
Armel TZ, Leinwand LA (2009) Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci U S A 106(15):6291–6296. https://doi.org/10.1073/pnas.0900107106
Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A (2003) Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 54(4):494–500. https://doi.org/10.1002/ana.10693
Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM, Hedera P, Fink JK, Petty EM, Lamont P, Fabian V, Bridges L, Voit T, Mastaglia FL, Laing NG (2004) Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 75(4):703–708. https://doi.org/10.1086/424760
Acknowledgements
We wish to thank the people with myopathy who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Ethical Committee of the Third Hospital of Hebei Medical University and was conducted in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, N., Zhao, Z., Shen, H. et al. MYH7 mutation associated with two phenotypes of myopathy. Neurol Sci 39, 333–339 (2018). https://doi.org/10.1007/s10072-017-3192-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-017-3192-2