
Abstract
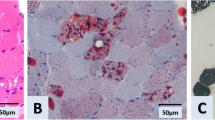
We report a case with late onset riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency (MADD) characterized by decreased acyl-carnitine profile in serum which is consistent with primary systemic carnitine deficiency (CDSP) while just the contrary to a typical MADD. This patient complained with muscle weakness, muscle pain and intermittent vomiting, and was diagnosed as polymyositis, received prednisone therapy before consulted with us. Muscle biopsy revealed mild lipid storage. The findings of serum acyl-carnitines were consistent with CDSP manifesting as decreased free and total carnitines in serum. But oral l-carnitine supplementation was not very effective to this patient and mutation analysis of the SLC22A5 gene for CDSP was normal. Later, another acyl-carnitine analysis revealed a typical MADD profile in serum, which was characterized by increased multiple acyl-carnitines. Compound heterozygous mutations were identified in electron transferring-flavoprotein dehydrogenase (ETFDH) gene which confirmed the diagnosis of MADD. After administration of riboflavin, he improved dramatically, both clinically and biochemically. Thus, late onset riboflavin-responsive MADD should be included in the differential diagnosis for adult carnitine deficiency.



Similar content being viewed by others

References
Frerman FE, Goodman SI (2001) Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric aciduria type 2. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds) The metabolic & molecular basis of inherited disease, 8th edn. McGraw-Hill, New York, pp 2357–2365
Olsen RKJ, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford MW, Dean JCS, Cornelius N, Andersen O, Oldfors A, Elisabeth H, Gregersen N, Turnbull DM, Morris AAM (2007) ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 130:2045–2054
Purevjav E, Kimura M, Takusa Y, Ohura T, Tsuchiya M, Hara N, Fukao T, Yamaguchi S (2002) Molecular study of electron transfer flavoprotein alpha-subunit deficiency in two Japanese children with different phenotypes of glutaric acidemia type II. Eur J Clin Invest 32:707–712
Wen B, Dai T, Li W, Zhao Y, Liu S, Zhang C, Li H, Wu J, Li D, Yan C (2010) Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry 81:231–236
Liang WC, Ohkuma A, Hayashi YK, Lopez LC, Hirano M, Nonaka I, Noguchi S, Chen LH, Jong YJ, Nishino I (2009) ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord 19:212–216
Wen B, Li D, Shan J, Liu S, Li W, Zhao Y, Lin P, Zheng J, Li D, Gong Y, Yan C (2013) Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol Genet Metab 109:154–160
Xi J, Wen B, Lin J, Zhu W, Luo S, Zhao C, Li D, Lin P, Lu J, Yan C (2014) Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 37: 399–404
DiMauro S, Garone C, Naini A (2010) Metabolic myopathies. Curr Rheumatol Rep 12:386–393
Kahler SG (2010) Metabolic Myopathies. In: Hoffmann GF, Zschocke J, Nyhan WL (eds) Inherited Metabolic Diseases. Berlin Heidelberg Springer-Verlag
Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, Guffon N, Besley GT, Onizuka R, De Meirleir LJ, Cvitanovic-Sojat L, Baric I, Dionisi-Vici C, Fumic K, Maradin M, Tein I (2002) Novel OCTN2 mutations: no genotype-phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am J Med Genet 111:271–284
Tang NL, Hui J, Law LK, To KF, Cheung KL, Magnus HN, Yuen PM, Fok TF (2000) Primary carnitine deficiency in the Chinese. Chin Med J (Engl) 113:376–380
Cruse RP, Di Mauro S, Towfighi J, Trevisan C (1984) Familial systemic carnitine deficiency. Arch Neurol 41:301–305
Izumi R, Suzuki N, Nagata M, Hasegawa T, Abe Y, Saito Y, Mochizuki H, Tateyama M, Aoki M (2011) A case of late onset riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency manifesting as recurrent rhabdomyolysis and acute renal failure. Int Med 50:2663–2668
Schimmenti LA, Crombez EA, Schwahn BC, Heese BA, Wood TC, Schroer RJ, Bentler K, Cederbaum S, Sarafoglou K, McCann M, Rinaldo P, Matern D, di San Filippo CA, Pasquali M, Berry SA, Longo N (2007) Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab 90:441–445
Magoulas PL, El-Hattab AW (2012) Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 7:68
El-Hattab AW, Li FY, Shen J, Powell BR, Bawle EV, Adams DJ, Wahl E, Kobori JA, Graham B, Scaglia F, Wong LJ (2010) Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med 12:19–24
Shibbani K, Fahed AC, Al-Shaar L, Arabi M, Nemer G, Bitar F, Majdalani M (2014) Primary carnitine deficiency: novel mutations and insights into the cardiac phenotype. Clin Genet 85:127–137
Shoji Y, Koizumi A, Kayo T, Ohata T, Takahashi T, Harada K, Takada G (1998) Evidence for linkage of human primary systemic carnitine deficiency with D5S436: a novel gene locus on chromosome 5q. Am J Hum Genet 63:101–108
Pestronk A Neuromuscular diseases center[online]. Available at: http://neuromuscular.wustl.edu/Neural. Accessed 2 Oct 2014
Xi J, Wen B, Lin J, Zhu W, Luo S, Zhao C, Li D, Lin P, Lu J, Yan C (2014) Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 37:399–404
Wang ZQ, Chen XJ, Murong SX, Wang N, Wu ZY (2011) Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J Mol Med (Berl) 89:569–576
Acknowledgments
We thank the patient and his family for their participation. This work was supported by the National Natural Science Foundation of China [Grant No. 81301071 and No. 81171182] and the Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP) [Grant No. 20130131120066].
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wen, B., Li, D., Li, W. et al. Multiple acyl-CoA dehydrogenation deficiency as decreased acyl-carnitine profile in serum. Neurol Sci 36, 853–859 (2015). https://doi.org/10.1007/s10072-015-2197-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-015-2197-y