
ABSTRACT
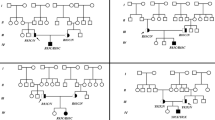
Glycogen storage disease type II (GSDII, Pompe's disease) is an autosomal recessive inherited deficiency of lysosomal α-glucosidase (GAA). Clinical as well as biochemical and allelic heterogeneity have been described in GSDII. We identified mutations within the GAA gene in seven unrelated German patients, six with adult- and one with juvenile-onset GSDII. Beside previously described mutations [IVS1 (–13T → G), Δexon 18, C1634T], we characterized four new mutations of GSDII: IVS6 (–22T → G), 271delG, G1912T (Gly638Trp), and 2432insC. The IVS6 (-22T → G) mutation gives rise to aberrant splicing, causing in-frame deletions of 25 or 40 amino acids within the GAA coding sequence and the insertion of a sequence of seven missense amino acids. Two affected siblings and an unrelated patient with adult GSDII are apparently homozygous for the exon 18 deletion. Both siblings are also heteroallelic for IVS1 (–13T → G). In conclusion, we observed pronounced allelic heterogeneity and an unexpectedly high frequency of homozygosity for larger in-frame deletions within the GAA coding sequence in German adult-onset GSDII patients.
Similar content being viewed by others


Author information
Authors and Affiliations
Additional information
Received: July 24, 1997 / Accepted: September 22, 1997
Rights and permissions
About this article
Cite this article
Vorgerd, M., Burwinkel, B., Reichmann, H. et al. Adult-onset glycogen storage disease type II: phenotypic and allelic heterogeneity in German patients. Neurogenetics 1, 205–211 (1998). https://doi.org/10.1007/s100480050030
Issue Date:
DOI: https://doi.org/10.1007/s100480050030