
Abstract
Early-onset Parkinson’s disease (EOPD) has been associated with recessive mutations in parkin (PARK2). About half of the mutations found in parkin are genomic rearrangements, i.e., large deletions or duplications. Although many different rearrangements have been found in parkin before, the exact breakpoints involving these rearrangements are rarely mapped. In the present study, the exact breakpoints of 13 different parkin deletions/duplications, detected in 13 patients out of a total screened sample of 116 EOPD patients using Multiple Ligation Probe Amplification (MLPA) analysis, were mapped using real time quantitative polymerase chain reaction (PCR), long-range PCR and sequence analysis. Deletion/duplication-specific PCR tests were developed as a rapid and low cost tool to confirm MLPA results and to test family members or patients with similar parkin deletions/duplications. Besides several different deletions, an exon 3 deletion, an exon 4 deletion and an exon 7 duplication were found in multiple families. Haplotype analysis in four families showed that a common haplotype of 1.2 Mb could be distinguished for the exon 7 duplication and a common haplotype of 6.3 Mb for the deletion of exon 4. These findings suggest common founder effects for distinct large rearrangements in parkin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder classically characterized by the clinical triad of bradykinesia, muscular rigidity and a resting tremor [1]. The mean age of onset for PD and early onset PD (EOPD) is 60 and 45 years, respectively [1, 2]. Autosomal recessive levodopa-responsive EOPD can be caused by mutations in parkin (PARK2), PINK1 or DJ-1 [2, 3]. Mutations in parkin are the most common among the recessive forms of PD [2]. This gene encodes Parkin, an E3 ubiquitin ligase. As part of the ubiquitin proteosome pathway, Parkin can protect neurons from cellular insults like alpha-synuclein toxicity and protosomal dysfunction [4]. Recent findings revealed that Parkin might also be involved in the development and/or progression of ovarian and other cancers, suggesting that parkin might also be a tumor suppressor gene [5, 6].
Since the cloning of parkin in 1998, a wide variety of mutations has been identified in this gene [7, 8]. About 50% of the mutations found in parkin are small (point) mutations detectable by sequencing. The remaining 50% consists of genomic rearrangements, i.e., large deletions or duplications [9, 10]. This high rearrangement rate can be explained by the fact that parkin is located within the large common fragile site (CFS) FRA6E [11]. CFS’s are reported to be highly susceptible to genomic instability [11]. Although little is known about the role of CFSs, many of the genes within these regions appear to function within the stress response system and in normal neurological development. The observation that extremely large genes, located in an unstable region, are highly evolutionary conserved suggests that these unstable regions and the genes within share a role in normal cell function [11].
Since genomic sequencing is not suitable to detect large rearrangements, other more quantitative methods are needed for a sensitive screening of these genomic variants. Available techniques include polymerase chain reaction (PCR)-based amplifications to detect homozygous deletions [3, 12], fluorescent dosage PCR [7], quantitative PCR (q-PCR) [8, 13–15], microarray [16] and Multiple Ligation Probe Amplification (MLPA) [17, 18]. MLPA is a rapid and sensitive technique to detect alterations in copy numbers using a multiplex PCR reaction. This method is suitable to identify deletions/duplications in parkin [17, 18]. Although many rearrangements have been found in parkin, the exact breakpoints involving these rearrangements are rarely mapped [3, 10, 12, 16]. In the present study, the breakpoint mapping of 13 different parkin deletions/duplications as found in 116 EOPD patients, is described. This approach enabled the development of specific deletion/duplication PCR’s as a rapid and low cost tool to confirm MLPA results and to test family members or patients with similar parkin deletions/duplications.
Materials and methods
Study population
The study population consisted of 116 patients whose samples were sent to the DNA diagnostic laboratory (Department of Clinical Genetics, Erasmus Medical Centre, Rotterdam, The Netherlands) between March 12, 1999 and February 3, 2010 for genetic screening of parkin. Samples were sent by neurologists (n = 89, 76.7%), clinical geneticists (n = 17, 14.6%) or other medical specialists (n = 10, 8.6%) because of a clinical diagnosis or symptoms of EOPD. The mean age of patients was 46.1 years (range 10–80 years) at time of blood sampling. The majority of the patients was male (n = 71, 61%).
Mutation analysis
DNA was extracted from peripheral blood using the DNA Blood Kit Special (Chemagen, Baesweiler, Germany), which is based on DNA extraction and purification with magnetic beads [19]. All 12 coding exons and exon/intron boundaries of parkin (NM_004562.2, NCBI build 36 (UCSC hg18, March 2006)) were screened using direct sequence analysis. SALSA MLPA kits P051 and P052 (MRC Holland, Amsterdam, The Netherlands) were used to detect large rearrangements in parkin. MLPA analysis was performed according to manufacturer’s instructions. MLPA products were run on an ABI 3730XL automated sequencer (Applied Biosystems, Foster City, CA, USA) and data was analyzed using Genemarker software version 1.5 (Softgenetics, State College, PA, USA).
q-PCR analysis
Real time quantitative PCR (q-PCR) was performed using Fam labelled Taqman assays, designed with Primer Express version 2.0.0 (Applied Biosystems), around the deleted/duplicated exons of parkin (NM_004562.2, NCBI build 36 (UCSC hg18, March 2006)). All primers were tested for specificity by performing a BLASTn run. Taqman probes were designed with an 8–10°C higher melting temperature (T m) than the primers. This was achieved by incorporating locked nucleic acid (LNA) monomers in the probe [20]. LNA containing probes have an enhanced hybridisation performance compared to classical Taqman probes and allow a shorter probe design [20]. T m calculations for LNA incorporated probes were performed using the Exiqon T m prediction tool (http://www.exiqon.com/ls/homeoflna/Oligo-tools/tm-prediction-tool.htm). LNA based Taqman assays were manufactured by Eurogentec (Seraing, Belgium). Gene dosage alterations were detected by performing a relative quantification run on an ABI7500 real-time PCR system (Applied Biosystems). Real-time PCR reactions were performed in a total volume of 25 μl, containing 20 ng DNA, 1× q-PCR mastermix Pluslow ROX (Eurogentec), 1× RNAse P (endogenous control) (Applied Biosystems), 30 μM forward and reverse primer and 10 μM probe. PCR conditions were initial 2 min 50°C incubation, followed by 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. All samples were analyzed in triplicate and compared to a normal control sample as calibrator [21].
Long-range PCR
Long-range PCR was performed using the Expand Long Template PCR System (Roche Applied Science, Indianapolis, IN, USA). PCR products were sequenced using the ABI 3730XL automated sequencer (Applied Biosystems). Data was analyzed using the Sequencing Analysis software version 5.3.1 (Applied Biosystems).
Deletion/duplication specific PCR
For all deletions/duplications, a specific deletion/duplication-PCR test was designed using two primers (primers A and B) flanking the deletion/duplication and one internal primer (primer C; also see Table 1). Obtained PCR products run on a 1.5% agarose gel will show a mutated allele and a normal allele for a patient with a heterozygous deletion/duplication, a mutated allele only for a patient with a homozygous deletion/duplication and a normal allele only for a non-affected individual.
Haplotype analysis
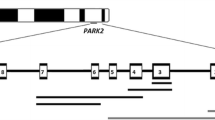
Haplotypes were constructed using nine polymorphic microsatellite markers around the parkin locus. Three markers were located within the intronic regions of parkin, the remaining six markers were located 5′ and 3′ of parkin on chromosome 6 (see Fig. 1). Markers were amplified using standard conditions and reagents. Fam labelled PCR products were run together with a LIZ-500 size standard on an ABI 3730XL automated sequencer (Applied Biosystems). Marker allele sizes were determined using Genemarker software version 1.5 (Softgenetics).

Schematic overview (not drawn to scale) of the polymorphic microsatellite markers used for haplotyping. On top of the bars the polymorphic microsatellite markers are depicted with their chromosomal marker positions in Mb according to NCBI build 36 (UCSC hg18, March 2006)
Results
A total of 116 patients with clinical symptoms of EOPD were screened for mutations in parkin using direct sequencing and MLPA analysis. In 13 patients (11%), at least one pathogenic mutation was found (24 parkin mutations were found in total). Eightythree percent (n = 20) of the pathogenic mutations consisted of large deletions/duplications, the remaining 17% (n = 4) consisted of point mutations or small deletions (e.g., a 2-bp deletion; data not shown). In Table 2, the characteristics of 12 families of the (index) patients with a large parkin deletion/duplication are summarized (families Tor-18-A and PV-24-A were reported before [22]). The remaining patient has one point mutation and was excluded from further analysis. Most of the patients with large deletions/duplications have classical parkinsonism syndromes like bradykinesia, hypokenetic rigidity and a resting tremor. The mean age of onset of the patients listed in Table 2 is 35.7 years. Table 3 gives an overview of all exon deletions and duplications found in our study population using MLPA analysis and breakpoint mapping.
To confirm the MLPA results with a different non-quantitative technique and to study recurrent deletions/duplications, the exact breakpoints of the rearrangements were mapped using q-PCR analysis; long-range PCR analysis and sequence analysis (see “Materials and methods” section).
Two examples of mapped rearrangements are shown in Fig. 2a (exon 4 deletion, Ex4del) and Fig. 2b (exon 7 duplication, Ex7dup). Ex4del also includes an insertion of 28 bp, whereas Ex7dup includes an insertion of 7 bp between the duplicated regions. The exact nomenclature of all deletions/duplications in this study is given in Table 3.

Schematic overview of the breakpoint analysis results of Ex4del (A) and Ex7dup (B). Retained sequences are shown in capitals, deleted nucleotides are shown in lower case, inserted nucleotides are in italic. Indicated for Ex7dup are the 5′ (vertically hatched) and 3′ flanking regions (horizontally hatched) that are duplicated together with exon 7, and the primers (a, b and c) used for the specific duplication PCR test (see “Materials and methods”). WT wild-type, dup duplication
For each exon deletion/duplication, a specific deletion/duplication PCR test was developed based on the breakpoint analysis results. Results of the specific deletion/duplication PCR tests are shown in Fig. 3. Most patients show a heterozygous pattern (both deletion/duplication band and wild-type band present), whereas for Ex5del and Ex8-10del a homozygous pattern was observed (only mutant band visible).

Results of all 13 deletion/duplication specific PCR tests for patients (P) versus CEPH controls (C). Samples were run on a 1.5% agarose gel together with a 1-kb DNA ladder (first lane; Invitrogen, Carlsbad, CA, USA). PCR products of the mutated alleles are smaller than PCR products of the normal alleles. Some background PCR bands are visible for Ex2dup (above normal band of 791 bp) and Ex2delA (below mutant band of 873 bp)
Most of the exon deletions/duplications appear to be unique, but three exon deletions/duplications (Ex3delB, Ex4del and Ex7dup) were found among multiple families (see Table 3). Three index patients had an exon 2 deletion (Table 3; families C, D and E), but the breakpoints were different. Three index patients had an exon 3 deletion of which two types were found (Table 3; families C, F and G). Four index patients had the same Ex7dup (Table 3; families D, H, I and J), whereas 2/4 index patients also shared the same Ex4del (Table 3; families H and I). All available family members of families D, H, I and J were screened with the specifically developed deletion/duplication PCR test. These screening results are shown in Fig. 4.

Four EOPD pedigrees with the same Ex7dup. Individuals with EOPD are depicted with filled symbols, half-filled symbols represent carriers of one mutated allele and open symbols represent individuals with an unknown PD history. Index patients are indicated with an arrow. Results of the deletion/duplication specific tests are given for each individual as −/−; no deletion/duplication detected or +/−; heterozygous deletion/duplication. Also indicated for each individual are the nine markers with corresponding allele sizes. Common haplotypes for Ex4del, Ex5-6del and Ex7dup are depicted as a black box, white box or grey box, respectively
Haplotypes were constructed around the parkin locus to investigate whether a common haplotype was present among the four families with the same Ex7dup mutation (Table 3; families D, H, I and J). Results of the haplotype analysis are presented in Fig. 4. For Ex7dup a shared haplotype was identified of at least 1.2 Mb as characterized by markers D6S1579 and D6S305. Ex4del shares a common haplotype of at least 6.3 Mb characterized by D6S955 to D6S386. Ex5-6del shares a common haplotype of at least 5.9 Mb characterized by D6S1708 to D6S305 found in one family only. Ex3del was found in two families (Table 3; families F and G), but for this mutation no common haplotype was found (data not shown).
Discussion
Over the past 11 years, 116 patients with early-onset Parkinson disease (EOPD) have been referred to our laboratory for genetic screening of parkin. Mutations were found in 13 patients with an overall mutation rate of 11%, which is slightly higher than reported before [14, 18, 23].
Multiplex Ligation-dependent Probe Amplification (MLPA) analysis was used in this study to screen for large deletions/duplications in parkin, because of its effectiveness to detect homo- and heterozygous deletions/duplications in parkin in a multiplex way. The MLPA technique is however prone to false positive results because of the possibility of polymorphisms on probe locations. Therefore, all deletions/duplications found need to be confirmed using a different technique [17, 23]. Our result that 83% of the mutations found in parkin consist of large deletions/duplications is higher than described previously [2, 14, 17, 18]. This finding clarifies that gene dosage studies are essential in parkin mutation screening. The high rate of large rearrangements might be explained by recurrent deletions/duplications in our study group or by the effectiveness of the MLPA technique to detect exon deletions/duplications compared to other techniques. Mapping of the exact breakpoints enables us to confirm MLPA results with a non-quantitative method and to compare the found deletions/duplications with other deletions/duplications in parkin. For all 13 different large deletions/duplications, a specific deletion/duplication-PCR test was developed as a low cost, quick, and robust technique to confirm MLPA results and to test other family members for the specific mutation or patients with similar MLPA results.
Only a few researchers have taken the effort to locate the exact breakpoints of large rearrangements found in parkin [3, 10, 12, 16], most likely because it is time-consuming as parkin is among the largest known genes with intronic regions up to 284 kb [11]. Four different exon 4 deletions were mapped before by two research groups [3, 10]. Ex4del in two of our patients (Fig. 2a) turned out to be the same deletion as reported by Hedrich et al. [10] in patients from Italy and Northern Germany. The compound heterozygous mutations of a deletion of exon 4 and a duplication of exon 7, as we found in families H and I, were reported previously in a Dutch patient [18]. In this patient, the reported mutations also occurred familial. Bayrakli et al. [16] mapped the breakpoints of an exon 5 deletion, but this deletion differs from the Ex5del found in our study. A duplication of exon 7 was reported by other researchers [7, 10, 18, 24–26], but was never mapped. Therefore, it remains undetermined whether these exon 7 duplications are identical. Most of the deletions/duplications listed in Table 3 are frameshift mutations that lead to a premature stopcodon several positions downstream. The only exception is Ex3-5del, which results in the loss of 149 residues. These residues are part of the Ubiquitin Like Domain (UBL) and the Src Homology 2 like (SH2-like) domain [6]. Although the exact function of these domains is still unclear, it is to be expected that the loss of 149 residues will lead to a loss of function of Parkin [6].
Similar haplotypes based on markers in the parkin region would support the hypothesis of a common founder effect multiple generations ago. The haplotype analysis (see Fig. 4) indicated that for Ex7dup a common haplotype of 1.2 Mb was present in four families, whereas a common haplotype of 6.3 Mb was present for Ex4del in two families. These data support the hypothesis of a common founder effect for mutations in parkin as reported before [10]. In autosomal recessive disease, founder mutations are commonly observed since in general there is no selection against carriers of mutations. Periquet et al. [27] reported a founder effect for some parkin point mutations, exon rearrangements were reported as independent recurrent events. It is unclear whether any of the exon rearrangements described by Periquet et al. are identical to ours since the deletions/duplications breakpoints were not mapped in their study.
A comprehensive literature overview among 379 unrelated carriers of parkin mutations showed a relatively high rate of exon deletions and duplications between exons 2 and 9 of parkin [10]. This is in line with the finding that the most unstable region in the CFS FRA6E was determined between exons 2 and 8 of parkin, suggesting hotspots for forming gaps and breaks in the large intronic regions [5]. In contradiction to this finding we found that in some cases (families D/H/I/J for Ex7dup; families F/G for Exdel3B and families H/I for Ex4del) deletions/duplications shared similar breakpoints (see Table 3), indicating a common founder effect. Our data provides evidence that, besides mutational hotspots, also common founder mutations are a source of exon rearrangements in parkin.
The parkin mutations found in pedigrees D, H and I follow a recessive inheritance pattern. In family J the difference between the affected patient II.4 and the unaffected individuals II.1 and II.2 who all share the same ex5-6del and are about the same age, remains unclear. The affected patient II.4 was negative for mutations in LRRK2, SNCA, PINK1 and DJ-1. It is possible that this patient has a second yet undiscovered mutation in a PD related gene. It is also possible that a single heterozygous mutation in a recessive gene like parkin is sufficient to cause PD or at least to increase the risk of developing the disease [2]. Individuals II.3 and II.5 of family J and individual I.1 of family I were unfortunately not available for study. For family D, only the index patient was available for testing.
In conclusion, with the development of multiplex screening techniques like MLPA, an increasing amount of rearrangements will be found in EOPD patients. In the present study we demonstrate that breakpoint mapping of parkin deletions/duplications can be useful to compare exon rearrangements between patients and families and to study a possible founder effect. Using this approach we have identified common haplotypes for an exon 7 duplication and an exon 4 deletion. Furthermore, we also show that deletion/duplication-specific PCR tests can be developed as a rapid and low cost tool to confirm MLPA results and to test family members or patients with similar parkin deletions/duplications.
References
Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Nicholl D, Breteler MMB, Oostra BA, De Mari M, Marconi R, Filla A, Bonnet A-M, Broussolle E, Pollak P, Rascol O, Rosier M, Arnould A, Brice A, French Parkinson’s Disease Genetics Study G, European Consortium on Genetic Susceptibility in Parkinson’s D (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342(21):1560–1567. doi:10.1056/NEJM200005253422103
Bonifati V (2007) Genetics of parkinsonism. Parkinsonism Relat Disord 13(Suppl 3):S233–S241
Clarimon J, Johnson J, Dogu O, Horta W, Khan N, Lees AJ, Hardy J, Singleton A (2005) Defining the ends of Parkin exon 4 deletions in two different families with Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 133B(1):120–123
von Coelln R, Dawson VL, Dawson TM (2004) Parkin-associated Parkinson's disease. Cell Tissue Res 318(1):175–184. doi:10.1007/s00441-004-0924-4
Denison SR, Wang F, Becker NA, Schule B, Kock N, Phillips LA, Klein C, Smith DI (2003) Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22(51):8370–8378
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA (2010) Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42(1):77–82. doi:ng.491[pii]10.1038/ng.491
Nichols WC, Pankratz N, Uniacke SK, Pauciulo MW, Halter C, Rudolph A, Conneally PM, Foroud T (2002) Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson's disease families. J Med Genet 39(7):489–492
Djarmati A, Hedrich K, Svetel M, Schafer N, Juric V, Vukosavic S, Hering R, Riess O, Romac S, Klein C, Kostic V (2004) Detection of Parkin (PARK2) and DJ1 (PARK7) mutations in early-onset Parkinson disease: Parkin mutation frequency depends on ethnic origin of patients. Hum Mutat 23(5):525. doi:10.1002/humu.9240
Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A (1999) A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet 8(4):567–574
Hedrich K, Eskelson C, Wilmot B, Marder K, Harris J, Garrels J, Meija-Santana H, Vieregge P, Jacobs H, Bressman SB, Lang AE, Kann M, Abbruzzese G, Martinelli P, Schwinger E, Ozelius LJ, Pramstaller PP, Klein C, Kramer P (2004) Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord 19(10):1146–1157. doi:10.1002/mds.20234
Smith DI, Zhu Y, McAvoy S, Kuhn R (2006) Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 232(1):48–57
Nakaso K, Adachi Y, Yasui K, Sakuma K, Nakashima K (2006) Detection of compound heterozygous deletions in the parkin gene of fibroblasts in patients with autosomal recessive hereditary parkinsonism (PARK2). Neurosci Lett 400(1–2):44–47. doi:S0304-3940(06)00167-4[pii]10.1016/j.neulet.2006.02.035
Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O, Schwinger E, Vieregge P, Lang AE, Breakefield XO, Ozelius LJ, Pramstaller PP, Klein C (2001) The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum Mol Genet 10(16):1649–1656
Kann M, Jacobs H, Mohrmann K, Schumacher K, Hedrich K, Garrels J, Wiegers K, Schwinger E, Pramstaller PP, Breakefield XO, Ozelius LJ, Vieregge P, Klein C (2002) Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 51(5):621–625. doi:10.1002/ana.10179
Periquet M, Latouche M, Lohmann E, Rawal N, De Michele G, Ricard S, Teive H, Fraix V, Vidailhet M, Nicholl D, Barone P, Wood NW, Raskin S, Deleuze JF, Agid Y, Durr A, Brice A (2003) Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 126(Pt 6):1271–1278
Bayrakli F, Bilguvar K, Mason CE, DiLuna ML, Bayri Y, Gungor L, Terzi M, Mane SM, Lifton RP, State MW, Gunel M (2007) Rapid identification of disease-causing mutations using copy number analysis within linkage intervals. Hum Mutat 28(12):1236–1240
Scarciolla O, Brancati F, Valente EM, Ferraris A, De Angelis MV, Valbonesi S, Garavaglia B, Uncini A, Palka G, Stuppia L, Dallapiccola B (2007) Multiplex ligation-dependent probe amplification assay for simultaneous detection of Parkinson's disease gene rearrangements. Mov Disord 22(15):2274–2278
Macedo MG, Verbaan D, Fang Y, van Rooden SM, Visser M, Anar B, Uras A, Groen JL, Rizzu P, van Hilten JJ, Heutink P (2009) Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson's disease. Mov Disord 24(2):196–203. doi:10.1002/mds.22287
Smit ML, Giesendorf BA, Heil SG, Vet JA, Trijbels FJ, Blom HJ (2000) Automated extraction and amplification of DNA from whole blood using a robotic workstation and an integrated thermocycler. Biotechnol Appl Biochem 32(Pt 2):121–125
Denys B, El Housni H, Nollet F, Verhasselt B, Philippe J (2010) A real-time polymerase chain reaction assay for rapid, sensitive, and specific quantification of the JAK2V617F mutation using a locked nucleic acid-modified oligonucleotide. J Mol Diagn 12(4):512–519. doi:jmoldx.2010.090137[pii]10.2353/jmoldx.2010.090137
Berggren P, Kumar R, Sakano S, Hemminki L, Wada T, Steineck G, Adolfsson J, Larsson P, Norming U, Wijkstrom H, Hemminki K (2003) Detecting homozygous deletions in the CDKN2A(p16(INK4a))/ARF(p14(ARF)) gene in urinary bladder cancer using real-time quantitative PCR. Clin Cancer Res 9(1):235–242
Bertoli-Avella AM, Giroud-Benitez JL, Akyol A, Barbosa E, Schaap O, van der Linde HC, Martignoni E, Lopiano L, Lamberti P, Fincati E, Antonini A, Stocchi F, Montagna P, Squitieri F, Marini P, Abbruzzese G, Fabbrini G, Marconi R, Dalla Libera A, Trianni G, Guidi M, De Gaetano A, Boff Maegawa G, De Leo A, Gallai V, de Rosa G, Vanacore N, Meco G, van Duijn CM, Oostra BA, Heutink P, Bonifati V (2005) Novel parkin mutations detected in patients with early-onset Parkinson's disease. Mov Disord 20(4):424–431
Keyser RJ, Lombard D, Veikondis R, Carr J, Bardien S (2009) Analysis of exon dosage using MLPA in South African Parkinson's disease patients. Neurogenetics. doi:10.1007/s10048-009-0229-6
Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC, Parkinson Study G (2003) Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 60(5):796–801
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31(7):763–780. doi:10.1002/humu.21277
Pramstaller PP, Kunig G, Leenders K, Kann M, Hedrich K, Vieregge P, Goetz CG, Klein C (2002) Parkin mutations in a patient with hemiparkinsonism–hemiatrophy: a clinical–genetic and PET study. Neurology 58(5):808–810
Periquet M, Lucking C, Vaughan J, Bonifati V, Durr A, De Michele G, Horstink M, Farrer M, Illarioshkin SN, Pollak P, Borg M, Brefel-Courbon C, Denefle P, Meco G, Gasser T, Breteler MM, Wood N, Agid Y, Brice A, French Parkinson's Disease Genetics Study Group. The European Consortium on Genetic Susceptibility in Parkinson's D (2001) Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am J Hum Genet 68(3):617–626. doi:S0002-9297(07)63102-4
Acknowledgements
The authors thank Raoul van de Graaf, Angelique Waals, Carola Jansen, Frank Magielsen, Marianne Deelen-Manders, Martine Parren-van Amelsvoort and Patricia Reichgelt of the DNA diagnostics laboratory, Clinical Genetics Department, Erasmus Medical Center in Rotterdam for screening the EOPD patients and Tom de Vries Lentsch of the Clinical Genetics Department of Erasmus Medical Center in Rotterdam for technical assistance with the figures.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Elfferich, P., Verleun-Mooijman, M.C., Maat-Kievit, J.A. et al. Breakpoint mapping of 13 large parkin deletions/duplications reveals an exon 4 deletion and an exon 7 duplication as founder mutations. Neurogenetics 12, 263–271 (2011). https://doi.org/10.1007/s10048-011-0302-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-011-0302-9