
Abstract
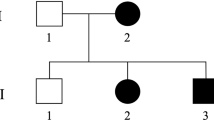
Spastic paraplegia type 4 (SPG4) is the most common autosomal dominant hereditary SPG caused by mutations in the SPAST gene. We studied the four-generation pedigree of a Japanese family with autosomal dominant hereditary SPG both clinically and genetically. Twelve available family members (ten affected; two unaffected) and two spouses were enrolled in the study. The clinical features were hyperreflexia in all four limbs, spasticity of the lower extremities, impaired vibration sense, mild cognitive impairment confirmed by the Wechsler Adult Intelligence Scale—Third Edition, and peripheral neuropathy confirmed by neurophysiological examinations. All four female patients experienced miscarriages. The cerebrospinal fluid tau levels were mildly increased in two of three patients examined. Linkage analyses revealed the highest logarithm of odds score of 2.64 at 2p23-p21 where the SPAST gene is located. Mutation scanning of the entire exonic regions of the SPAST gene by direct sequencing revealed no mutations. Exonic copy number analysis by real-time quantitative polymerase chain reaction revealed heterozygous deletion of exons 1 to 4 of the SPAST gene. Breakpoint analysis showed that the centromeric breakpoint was located within intron 4 of SPAST while the telomeric breakpoint was located within intron 3 of the neighboring DPY30 gene, causing a deletion of approximately 70 kb ranging from exons 1 to 3 of DPY30 to exons 1 to 4 of SPAST. To our knowledge, this is the first report of SPG4 associated with partial deletions of both the SPAST and DPY30 genes. The partial heterozygous deletion of DPY30 could modify the phenotypic expression of SPG4 patients with this pedigree.



Similar content being viewed by others

References
Dürr A, Davoine CS, Paternotte C, von Fellenberg J, Cogilnicean S, Coutinho P et al (1996) Phenotype of autosomal dominant spastic paraplegia linked to chromosome 2. Brain 119:1487–1496
Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F et al (1999) Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 23:296–303
Errico A, Ballabio A, Rugarli EI (2002) Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet 11:153–163
Roll-Mecak A, Vale RD (2008) Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451:363–367. doi:10.1038/nature06482
Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP (2005) Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol 168:599–606. doi:10.1083/jcb.200409058
Salinas S, Carazo-Salas RE, Proukakis C, Cooper JM, Weston AE, Schiavo G et al (2005) Human spastin has multiple microtubule-related functions. J Neurochem 95:1411–1420. doi:10.1111/j.1471-4159.2005.03742.x
Iwanaga H, Tsujino A, Shirabe S, Eguchi H, Fukushima N, Niikawa N et al (2005) Large deletion involving the 5′-UTR in the spastin gene caused mild phenotype of autosomal dominant hereditary spastic paraplegia. Am J Med Genet A 133:13–17. doi:10.1002/ajmg.a.30510
Beetz C, Nygren AOH, Schickel J, Auer-Grumbach M, Bürk K, Heide G et al (2006) High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology 67:1926–1930
Depienne C, Fedirko E, Forlani S, Cazeneuve C, Ribaï P, Feki I et al (2007) Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J Med Genet 44:281–284. doi:10.1136/jmg.2006.046425
Svenstrup K, Bross P, Koefoed P, Hjermind LE, Eiberg H, Born AP et al (2009) Sequence variants in SPAST, SPG3A and HSPD1 in hereditary spastic paraplegia. J Neurol Sci 284:90–95. doi:10.1016/j.jns.2009.04.024
Erichsen AK, Inderhaug E, Mattingsdal M, Eiklid K, Tallaksen CME (2007) Seven novel mutations and four exon deletions in a collection of Norwegian patients with SPG4 hereditary spastic paraplegia. Eur J Neurol 14:809–814. doi:10.1111/j.1468-1331.2007.01861.x
Fonknechten N, Mavel D, Byrne P, Davoine CS, Cruaud C, Boentsch D et al (2000) Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet 9:637–644
White KD, Ince PG, Lusher M, Lindsey J, Cookson M, Bashir R et al (2000) Clinical and pathologic findings in hereditary spastic paraparesis with spastin mutation. Neurology 55:89–94
Lindsey JC, Lusher ME, McDermott CJ, White KD, Reid E, Rubinsztein DC et al (2000) Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet 37:759–765
Tallaksen CME, Guichart-Gomez E, Verpillat P, Hahn-Barma V, Ruberg M, Fontaine B et al (2003) Subtle cognitive impairment but no dementia in patients with spastin mutations. Arch Neurol 60:1113–1118
Ribaï P, Depienne C, Fedirko E, Jothy A-C, Viveweger C, Hahn-Barma V et al (2008) Mental deficiency in three families with SPG4 spastic paraplegia. Eur J Hum Genet 16:97–104. doi:10.1038/sj.ejhg.5201922
McDermott CJ, Burness CE, Kirby J, Cox LE, Rao DG, Hewamadduma C et al (2006) Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology 67:45–51
Crippa F, Panzeri C, Martinuzzi A, Arnoldi A, Redaelli F, Tonelli A et al (2006) Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia. Arch Neurol 63:750–755
Mitne-Neto M, Kok F, Beetz C, Pessoa A, Bueno C, Graciani Z et al (2007) A multi-exonic SPG4 duplication underlies sex-dependent penetrance of hereditary spastic paraplegia in a large Brazilian pedigree. Eur J Hum Genet 15:1276–1279
Magariello A, Muglia M, Patitucci A, Ungaro C, Mazzei R, Gabriele AL et al (2010) Mutation analysis of the SPG4 gene in Italian patients with pure and complicated forms of spastic paraplegia. J Neurol Sci 288:96–100. doi:10.1016/j.jns.2009.09.025
Murphy S, Gorman G, Beetz C, Byrne P, Dytko M, McMonagle P et al (2009) Dementia in SPG4 hereditary spastic paraplegia: clinical, genetic, and neuropathologic evidence. Neurology 73:378–384
McMonagle P, Byrne PC, Fitzgerald B, Webb S, Parfrey NA, Hutchinson M (2000) Phenotype of AD-HSP due to mutations in the SPAST gene: comparison with AD-HSP without mutations. Neurology 55:1794–1800
Orlacchio A, Kawarai T, Gaudiello F, Totaro A, Schillaci O, Stefani A et al (2005) Clinical and genetic study of a large SPG4 Italian family. Mov Disord 20:1055–1059. doi:10.1002/mds.20494
Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DVK et al (2009) Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet 17:187–194. doi:10.1038/ejhg.2008.147
Nielsen JE, Johnsen B, Koefoed P, Scheuer KH, Grønbech-Jensen M, Law I et al (2004) Hereditary spastic paraplegia with cerebellar ataxia: a complex phenotype associated with a new SPG4 gene mutation. Eur J Neurol 11:817–824
Chinnery PF, Keers SM, Holden MJ, Ramesh V, Dalton A (2004) Infantile hereditary spastic paraparesis due to codominant mutations in the spastin gene. Neurology 63:710–712
Miura S, Shibata H, Kida H, Noda K, Tomiyasu K, Yamamoto K et al (2008) Hereditary motor and sensory neuropathy with proximal dominancy in the lower extremities, urinary disturbance, and paroxysmal dry cough. J Neurol Sci 273:88–92. doi:10.1016/j.jns.2008.06.027
Wharton S, McDermott CJ, Grierson AJ, Wood JD, Gelsthorpe C, Ince PG et al (2003) The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J Neuropathol Exp Neurol 62:1166–1177
Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101:1371–1378
Georgieff IS, Liem RKH, Mellado W, Nunez J, Shelanski ML (1991) High molecular weight tau: preferential localization in the peripheral nervous system. J Cell Sci 100:55–60
Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW et al (2009) MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology 73:294–301
Nielsen JE, Jennum P, Fenger K, Sørensen SA, Fuglsang-Frederiksen A (2001) Increased intracortical facilitation in patients with autosomal dominant pure spastic paraplegia linked to chromosome 2p. Eur J Neurol 8:335–339
Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis disease. Hum Mol Genet 13:R57–R64. doi:10.1093/hmg/ddh073
Cho Y-W, Hong T, Hong SH, Guo H, Yu H, Kim D et al (2007) PTIP associates with MLL3- and MLL4- containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406. doi:10.1074/jbc.M701574200
Hsu DR, Meyer BJ (1994) The dpy-30 gene encodes an essential component of the Caenorhabditis elegans dosage compensation machinery. Genetics 137:999–1018
Acknowledgments
The authors thank all of the individuals for their participation in this study.
Conflicts of interest
The authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miura, S., Shibata, H., Kida, H. et al. Partial SPAST and DPY30 deletions in a Japanese spastic paraplegia type 4 family. Neurogenetics 12, 25–31 (2011). https://doi.org/10.1007/s10048-010-0260-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-010-0260-7