
Abstract
Mutations in the ATP13A2 (PARK9) and FBXO7 (PARK15) genes are linked to different forms of autosomal recessive juvenile-onset neurodegenerative diseases with overlapping phenotypes, including levodopa-responsive parkinsonism, pyramidal disturbances, cognitive decline, and supranuclear gaze disturbance. However, the associated genotypes and phenotypes are poorly characterized due to the small number of patients described. Here, we report clinical, instrumental, and genetic findings in an Italian family with novel PARK9 and PARK15 mutations. The proband developed a severe progressive phenotype including juvenile-onset parkinsonism, pyramidal disturbances, cognitive decline, and oculomotor abnormalities. On the contrary, his brother only shows mild abnormalities (pyramidal, cognitive, and oculomotor) on the neurological examination at the age of 31 years. These two brothers both carry a novel homozygous PARK9 missense (p.G877R) and a novel heterozygous PARK15 mutation (p.R481C). The PARK9 mutation replaces a crucial residue for the ATPase activity, and is therefore most likely a loss-of-function mutation and disease-causing in homozygous state. The pathogenic significance of the PARK15 single heterozygous mutation remains unclear. In both sibs, DaTSCAN single photon emission computed tomography showed marked nigrostriatal dopaminergic defects, and transcranial magnetic stimulation detected prolonged central motor conduction time. MRI, including T2*-weighted imaging, detected no evidence of brain iron accumulation. This family, the third reported with homozygous PARK9 mutations and the first with mutations in two genes for atypical juvenile parkinsonism, illustrates that PARK9-linked disease might display wide intra-familial clinical variability and milder phenotypes, suggesting the existence of strong, still unknown, modifiers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
PARK9, or Kufor-Rakeb syndrome (KRS), is characterized by juvenile-onset, levodopa-responsive parkinsonism, pyramidal signs, dementia, and supranuclear gaze palsy [1, 2], caused by recessive mutations in the ATP13A2 gene [3]. Since the initial report, only six definitely disease-causing (homozygous or compound heterozygous) mutations have been described in two families [3] and three isolated KRS patients [4–6]. For another seven mutations identified in single heterozygous state in patients with early-onset Parkinson’s disease (PD) [4, 7, 8], the role in the disease causation remains unclear.
The ATPA13A2 gene encodes a large lysosomal transmembrane protein, belonging to the group 5 P-type ATPase family. P-type ATPases use the energy deriving from ATP hydrolysis to generate gradients of ions across membranes [9]. The substrate specificity of the ATP13A2 protein remains unknown, but there is evidence that this protein is involved in the transport of several cations from the cytosol to the lysosomal lumen, including manganese, cadmium, nickel, and selenium [10, 11].
Recessive mutations in the FBXO7 gene have been recently linked to PARK15, a different rare juvenile neurodegenerative disorder, presenting as a parkinsonian-pyramidal syndrome and reported so far in only three families [12, 13].
In this study, we describe two brothers from a southern Italian family, who both carry a novel homozygous ATP13A2 mutation and a single heterozygous FBXO7 mutation. Despite the identical genotypes, the brothers were affected with markedly different severity.
Subjects and methods
A simplified pedigree is shown in Fig. 1. A healthy couple had two offspring, who both developed different degrees of a progressive neurological disease. The parents report no known consanguinity, but they both originate from the same small village in the Campania region of southern Italy. The project was approved by the relevant ethical authorities, and written informed consent was obtained from all subjects.

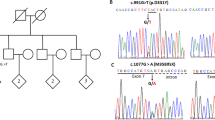
Genetic findings in the family with PARK9 and PARK15 mutations. a Electropherograms of a fragment of ATP13A2 (PARK9) exon 24; hom homozygous mutation, het heterozygous mutation, wt wild-type sequence. b Simplified pedigree. The proband is highlighted by a black symbol; his brother is highlighted in gray. The ATP13A2 (PARK9) and FBXO7 (PARK15) mutations are shown below the individual symbols. c Electropherograms of a fragment of FBXO7 (PARK15) exon 9; het heterozygous mutation, wt wild-type sequence. d Schematic representation of the ATP13A2 protein, its known functional domains and amino acid motifs. The position of the G877R mutation detected in the Italian family is indicated within the P2 domain and within the MCGDG motif (the mutated G is underlined). The other two missense mutations reported previously in homozygous state in PARK9 patients are also shown. e Schematic representation of the FBXO7 protein. The R481C mutation detected in the Italian family is framed. The other disease-causing mutations reported previously in PARK15 patients are also shown. f Alignment of ATP13A2 protein homologues in the region of the G877R mutation. g Alignment of P5-ATPase protein family in the region corresponding to the G877R mutation. h Alignment of FBXO7 protein homologues in the region of the R481C mutation
Genetic studies
Genomic DNA was isolated from peripheral blood samples following standard procedures. We first screened the proband for mutations in the parkin (PARK2), PINK1 (PARK6), DJ-1 (PARK7), ATP13A2 (PARK9), PLA2G6 (PARK14), and FBXO7 (PARK15) genes. All exons and exon–intron boundaries of the above-mentioned genes were sequenced in both strands. The PCR protocols and primers are available on request from the authors. Direct sequencing was performed using Big Dye Terminator chemistry ver.3.1 (Applied Biosystems). Fragments were loaded on an Applied Biosystems 3130XL automated sequencer and analyzed with DNA Sequencing Analysis (ver.5.3) and SeqScape (ver.2.6) software (Applied Biosystems). The mutations were numbered from the “A” of the ATG-translation initiation codon, according to the GenBank reference sequences, accession numbers NM_022089.2 (ATP13A2) and NM_012179.3 (FBXO7). We also screened the proband for copy number aberrations in the parkin, PINK1, DJ-1, ATP13A2, and SNCA (alpha-synuclein) genes, using a multiplex ligation-dependent probe amplification (MLPA) assay (probe mix P051 and P052) according to the manufacturer’s protocol (MRC-Holland). The PARK9 and PARK15 mutations detected in the proband were studied in the whole family, as well as in a panel of unrelated healthy controls from the same region of southern Italy, and using the same sequencing techniques. In order to analyze the evolutionary conservation of the mutated amino acids, the ATP13A2 and FBXO7 protein homologues were aligned using the program ClustalW.
Brain imaging
Brain single photon emission computed tomography (SPECT) studies were performed 4 h after intravenous administration of 185 MBq of [123I]FP-CIT (DaTSCAN, GE-Healthcare) using a dual-headed camera (E.CAM, Siemens Medical Systems, Hoffman Estates, IL) equipped with low-energy high-resolution collimators (zoom, 1.23; pixel size, 3.90 × 3.90 mm). Outcome measures were the specific-to-nondisplaceable binding ratio, V″3 for the putamen and caudate.
The brain MRI studies included T2-weighted turbo spin-echo (TSE) (TR/TE 4,400/100 ms) and FLAIR (TR/TE/TI 8,000/100/2,200 ms) sequences, as well as T1-weighted conventional SE (TR/TE 580/15 ms), and DWI (EPI, TR/TE 3,500/90 ms) images (1.5 T, Achieva, Philips Medical Systems, Eindhoven, Netherlands). In addition, T2*-weighted axial images (Gradient echo, TR/TE 600/15 ms, Flip Angle 20°) were acquired (3 T, Magnetom Trio, Siemens Medical Systems, Erlangen, Germany).
Results
Clinical findings
The proband (NAPO-6 in Fig. 1) is a 41-year-old man with referred perinatal asphyxia and slight delay in reaching the developmental milestones. At the age of 10 years, he developed a slowly progressive gait disturbance with slowing and stiffness of legs. An asymmetric onset of symptoms was not recorded. Treatment with levodopa yielded marked improvement in gait and in limb rigidity. Nonetheless, over time, his motor performance progressively deteriorated, and a mild cognitive impairment became apparent. At the age of 18 year, he received the diagnosis of “extra-pyramidal and pyramidal syndrome with cognitive impairment”. Since then, the patient complained of dysarthria and dysphagia, and some years later, the arm and leg stiffness became so severe to determine the complete dependence on others for all daily living activities. Our neurological examination, 30 years after the disease onset, showed a moderate limitation of the up-gaze and right lateral gaze. The saccadic eye movements were slow both in upward and downward direction and bilaterally hypometric on lateral gaze. A severe hypomimia was present, and the speech was almost incomprehensible. There was also mild dysphagia, allowing a normal feeding. Sub-continuous mini-myoclonus was present in the lower facial muscles. The gait was only possible with bilateral support, and dystonic posturing was present. The muscle tone was markedly increased in all limbs with prevalence of rigidity in the arms and spasticity in the legs. The deep tendon reflexes were all brisk, and the Babinski sign and palmomental reflex were present bilaterally. Cerebellar signs and tremor were absent. The unified PD rating scale (UPDRS) motor score was 67, and it was not modified by the acute administration of levodopa. The administration of the mini-mental state examination (MMSE) was not possible. Blood chemistry, including manganese, selenium, cadmium, and nickel dosage, was normal. Needle EMG (biceps brachii, rectus femori, and tibialis anterior muscles), surface antidromic sensory (median, ulnar, sural, and peroneal nerves) and orthodromic motor nerve (median, ulnar, and tibial nerves) conduction studies (NCS), and somatosensory evoked potentials (SSEPs) following median and tibial nerve were all normal. Transcranial Magnetic Stimulation (TMS) showed a prolonged central motor conduction time (CMCT) for upper (10.3 ms; normal upper limit, 8 ms) and lower (30.5 ms; normal upper limit, 15 ms) limbs, while motor evoked potential (MEP) amplitude was normal in the upper and reduced in the lower limbs (0.7 mV; normal lower limit, 1.2 mV).
The proband’s brother (NAPO-7 in Fig. 1), a 31-year-old man, had a normal birth and developmental motor milestones, but he did not perform normally at school and received a diagnosis of “mild mental retardation”. At the time of neurological examination (age 31), he was otherwise asymptomatic. However, the neurological evaluation showed a slight limitation of upward and downward gaze, moderate increase of axial tone, and slight upper limb rigidity. The deep tendon reflexes were brisk, and the Babinski sign was bilaterally present. Finger tapping and stepping were moderately decreased in frequency and amplitude. Cerebellar signs and tremor were absent. The UPDRS motor score was 16. He had never used antiparkinsonian drugs. The MMSE score was 23. Blood chemistry, including manganese, selenium, cadmium, and nickel dosage, was normal. Needle EMG (biceps brachii, rectus femori, and tibialis anterior muscles), surface antidromic sensory (median, ulnar, sural, and peroneal nerves) and orthodromic motor (median, ulnar, and tibial nerves) NCS, and SSEPs following median and tibial nerve were normal. TMS showed a prolonged CMCT for upper (11.8 ms; normal upper limit, 8 ms) and lower (24.2 ms; normal upper limit, 15 ms) limbs, while MEP amplitude was normal in the upper and reduced in the lower limbs (0.4 mV; normal lower limit, 1.2 mV).
Genetic findings
By sequencing the genomic DNA in the proband, we detected a homozygous mutation in exon 24 of the ATP13A2 gene (c.G2629A, predicted to lead to the p.G877R missense change in the encoded protein) and a heterozygous mutation in exon 9 of the FBXO7 gene (c.C1441T, predicted to result in the missense change p.R481C) (Fig. 1a–c). Direct sequencing revealed no additional mutations in these or other screened genes for autosomal recessive parkinsonism (parkin, PINK1, DJ-1, PLA2G6). Furthermore, gene dosage aberrations were not detected by the MLPA assays in any of the screened genes. The ATP13A2 c.G2629A mutation was detected in heterozygous state in both the proband’s parents, while it was absent from a large number of control chromosomes (n = 336) from the same region of southern Italy. The FBXO7 c.C1441T mutation was detected in heterozygous state in the proband’s mother, while it was absent from the father and from control chromosomes from the region (n = 318). The proband’s brother (NAPO-7) carried a homozygous ATP13A2 and heterozygous FBXO7 mutation, a situation identical to that found in the proband (Fig. 1b). The cDNA studies confirmed the genotypes detected in genomic DNA in the whole family (not shown). Both mutations introduce drastic replacements in amino acid positions that have been highly conserved in the evolution of the ATP13A2 and FBXO7 protein homologues (Fig. 1d–h).
Imaging findings
The [123I]FP-CIT–SPECT showed in both brothers a decrease of dopamine transporter (DaT) density in the striatum (Fig. 2). Compared to controls, the proband showed a marked and widespread striatal reduction of V″3 (75% in the caudate and 85% in the putamen) (Fig. 2a). In the brother, the specific-to-nondisplaceable binding ratio V″3 was also symmetrically reduced, mostly in the putamen (40% in the caudate and 65% in the putamen) (Fig. 2b). In both brothers the brain MRI showed diffuse cerebral and cerebellar cortex atrophy, which was more severe in the proband (Fig. 3). The T2* scans detected no evidence of metal accumulation in the basal ganglia of both sibs (Fig. 3c, f).

DaTSCAN SPECT imaging. Brain [123I]FP-CIT (DaTSCAN SPECT) in the proband (a), his brother (b), and a normal unrelated subject (c); a severe presynaptic defect of the nigrostriatal dopaminergic systems is present in the two brothers, more marked in the proband

Brain MRI imaging. T2-weighted TSE axial scans (1.5 T) at the level of the cerebellar hemispheres (a, d) and centra semiovalia (b, e) and T2*-weighted scans (3 T) at the level of the basal ganglia (c, f), in the proband (a–c) and his brother (d–f). Diffuse supra- and sub-tentorial atrophy is present. The T2*-weighted scans show normal intensity of the basal ganglia in both individuals. Incidental left maxillary sinusitis is also present (d)
Discussion
Several arguments support the view that the G877R mutation is harmful for the ATP13A2 protein function. The glycine 877 lies in a stretch of five amino acids that have not only been conserved in the evolution of ATP13A2 homologues until the yeasts but are also invariably present in all members of the ATPase P5 protein family (Fig. 1f–g), suggesting a crucial role for the protein function. Furthermore, the G877R mutation replaces a small non-polar amino acid (glycine) with a larger polar one (arginine). The large cytoplasmic loop located between the M4 and M5 transmembrane domains of the ATP13A2 protein (Fig. 1d) contains the catalytic autophosphorylation domain (P domain, including an N-terminal and C-terminal part) and the nucleotide binding domain (N domain, necessary for ATP binding) [9, 14]. The presence of an arginine in position 877 places a net positive charge next to the highly conserved negatively charged aspartic acid in position 878, in a very important motif within the C-terminal part of the P domain (Fig. 1d).
Structural studies of a similar P-type cationic pump, the calcium ATPase of skeletal muscle sarcoplasmic reticulum (SERCA1), revealed that the conserved aspartic acid corresponding to Asp-878 in ATP13A2 is one of the most critical residues for the ATP hydrolysis and is spatially very close to the aspartic acid residue (Asp351, corresponding to Asp513 in ATP13A2) which provides the autophosphorylation site [14]. Thus, the G877R mutation is very likely to interfere with the ATPase and autophosphorylation activity of ATP13A2, necessary for the function as cationic pump. Taken together, these considerations argue strongly that the G877R mutation is deleterious for the ATP13A2 function and disease-causing when present in homozygous state.
The R481C mutation replaces a highly conserved arginine residue in the FBXO7 protein, within the so called R(Ar)DP motif (where Ar indicates any aromatic amino acid) in the C-terminal proline-rich region of the protein (Fig. 1e, h) [15]. The function of the R(Ar)DP motif remains undetermined, but the proline-rich region is important for binding of the known FBXO7 substrates [15–17]. The mutation might therefore also be deleterious for the FBXO7 function. However, being found as a single heterozygous mutation in the two affected brothers and their unaffected mother, its significance for the disease causation remains unclear. This could be a coincidental finding, or the mutation could act as a disease modifier, or play some pathogenic role, together with still unknown mutations in other genes.
Different research lines point to an important role for the lysosomes in the pathogenesis of common, late-onset PD. Lysosomes are important for the degradation of the alpha-synuclein protein [18], and heterozygous mutations in the GBA gene, encoding the lysosomal enzyme glucocerebrosidase, are an important risk factor for PD [19]. Intriguingly, the ATP13A2 mRNA is highly expressed in the brain, particularly in substantia nigra, and it might be up-regulated in the brain of patients with the common late-onset idiopathic PD [3]. Moreover, the ATP13A2 protein has been recently identified as a potent modifier of the toxicity induced by alpha-synuclein in animal models of PD [10]. On the other hand, manganese, one possible substrate of ATP13A2, is a well-known cause of toxic parkinsonism in humans [20]. Despite the rarity of PARK9 mutations, the ATP13A2 protein might therefore offer clues for understanding the pathogenesis of the common, late-onset forms of PD, linking genetic (alpha-synuclein), and environmental (manganese) factors in the disease etiology.
The clinical phenotype associated with PARK9 mutations remains poorly defined, as only few patients with clear disease-causing genotypes have been reported. Some phenotypic variability has been described (i.e., variable presence and degrees of dementia, behavior disorders, visual hallucinations, l-dopa responsiveness, and disease progression). Our proband resembles indeed the previously reported PARK9 cases, and his phenotype is compatible with the clinical diagnosis of KRS. The sub-continuous mini-myoclonus present in the lower facial muscles is reminiscent of the facial–faucial–finger mini-myoclonus described in the original KRS patients [2] and other PARK9 cases [3, 5]. Remarkably, the younger brother, apart from a moderate cognitive deficit, remains otherwise asymptomatic at the age of 31 years, and only the neurological examination revealed a mild pyramidal–extra-pyramidal involvement. This marked phenotypic variability has not been previously described in patients with identical PARK9 mutations. It could be the result of the action of genetic or environmental modifiers, or both. An aggravating effect for the perinatal brain sufferance in the proband could not be excluded; however, the severity of his phenotype is similar to that in the other PARK9 patients, and therefore, a protective effect in the proband’s brother appears more likely. Mutations in the other known genes for early-onset PD (apart from the FBXO7 heterozygous mutation) were excluded in the proband, and therefore they cannot play a role as aggravating factors. Environmental factors could include the role of some cations, according to the evidence that the ATP13A2 protein is involved in their transport from the cytosol to the lysosomal lumen [10, 11]. However, the blood levels of manganese, cadmium, nickel, and selenium were normal in both sibs. Furthermore, the siblings have been living with their parents in the same place, being likely exposed to the same environment.
In both sibs, the brain MRI showed diffuse brain atrophy, more marked in the proband, and the DaTSCAN SPECT showed marked nigrostriatal dopaminergic defects, again more marked in the proband, and similar to those reported in advanced idiopathic PD. The neurophysiologic investigation confirmed the involvement of the pyramidal tract and showed the normality of the large myelinated peripheral fibers and of the somatosensory system.
Recently, hypointense signals in the basal ganglia in MRI T2*-weighted scans, suggesting iron accumulation, have been reported in a patient with homozygous PARK9 mutation [6]. Based on this finding, it has been suggested to classify PARK9 within the neurodegenerations with brain iron accumulation [6]. The MRI studies in other previously reported PARK9 cases did not include T2*-weighted scans [2, 5] and were therefore inconclusive concerning the iron accumulation. Using a comprehensive MRI protocol that includes T2*-weighted scans, we did not find evidence of metal accumulation in the basal ganglia of both sibs reported here. Brain metal accumulation seems therefore not a constant feature in PARK9, even after three decades of disease course. Perhaps brain metal accumulation occurs in the cases with ATP13A2 mutations leading to the most severe loss of protein function, such as that present in the patient reported elsewhere [6] (p.Thr367ArgfsX29, causing frameshift and protein truncation), while in some patients with missense mutations a residual protein function is retained and brain metal accumulation does not occur.
In conclusion, this family, the third reported with homozygous PARK9 mutations and the first with mutations in two genes for atypical juvenile parkinsonism, illustrates that PARK9-linked disease might display wide intra-familial clinical variability and present with milder phenotypes, suggesting the existence of strong, still unknown, modifiers.
References
Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M (1994) Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 89:347–352
Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ (2005) Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 20:1264–1271
Ramirez A, Heimbach A, Grundemann J et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191
Di Fonzo A, Chien HF, Socal M et al (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68:1557–1562
Ning YP, Kanai K, Tomiyama H et al (2008) PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 70:1491–1493
Schneider SA, Paisan-Ruiz C, Quinn NP et al (2010) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 25:979–984
Lin CH, Tan EK, Chen ML et al (2008) Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 71:1727–1732
Djarmati A, Hagenah J, Reetz K et al (2009) ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord 24:2104–2111
Schultheis PJ, Hagen TT, O’Toole KK et al (2004) Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochem Biophys Res Commun 323:731–738
Gitler AD, Chesi A, Geddie ML et al (2009) Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41:308–315
Schmidt K, Wolfe DM, Stiller B, Pearce DA (2009) Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun 383:198–202
Shojaee S, Sina F, Banihosseini SS et al (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82:1375–1384
Di Fonzo A, Dekker MC, Montagna P et al (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72:240–245
Toyoshima C, Nakasako M, Nomura H, Ogawa H (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405:647–655
Kirk R, Laman H, Knowles PP et al (2008) Structure of a conserved dimerization domain within the F-box protein Fbxo7 and the PI31 proteasome inhibitor. J Biol Chem 283:22325–22335
Hsu JM, Lee YC, Yu CT, Huang CY (2004) Fbx7 functions in the SCF complex regulating Cdk1-cyclin B-phosphorylated hepatoma up-regulated protein (HURP) proteolysis by a proline-rich region. J Biol Chem 279:32592–32602
Chang YF, Cheng CM, Chang LK, Jong YJ, Yuo CY (2006) The F-box protein Fbxo7 interacts with human inhibitor of apoptosis protein cIAP1 and promotes cIAP1 ubiquitination. Biochem Biophys Res Commun 342:1022–1026
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Pal PK, Samii A, Calne DB (1999) Manganese neurotoxicity: a review of clinical features, imaging and pathology. Neurotoxicology 20:227–238
Acknowledgements
This study was supported by a grant from the Internationaal Parkinson Fonds (The Netherlands) to Dr. Bonifati. We thank all the family members for their participation and Tom de Vries-Lentsch (Department of Clinical Genetics, Erasmus MC) for the artwork.
Disclosure
All experiments comply with the current laws of the country in which they were performed. The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Santoro, L., Breedveld, G.J., Manganelli, F. et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 12, 33–39 (2011). https://doi.org/10.1007/s10048-010-0259-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-010-0259-0