
Abstract
Autosomal recessive hereditary spastic paraplegia with thin corpus callosum (HSP-TCC) maps to the SPG11 locus in the majority of cases. Mutations in the KIAA1840 gene, encoding spatacsin, have been shown to underlie SPG11-linked HSP-TCC. The aim of this study was to perform candidate gene analysis in HSP-TCC subjects from Asian families and to characterize disruption of spatacsin function during zebrafish development. Homozygosity mapping and direct sequencing were used to assess the ACCPN, SPG11, and SPG21 loci in four inbred kindreds originating from the Indian subcontinent. Four novel homozygous SPG11 mutations (c.442+1G>A, c.2146C>T, c.3602_3603delAT, and c.4846C>T) were identified, predicting a loss of spatacsin function in each case. To investigate the role of spatacsin during development, we additionally ascertained the complete zebrafish spg11 ortholog by reverse transcriptase PCR and 5′ RACE. Analysis of transcript expression through whole-mount in situ hybridization demonstrated ubiquitous distribution, with highest levels detected in the brain. Morpholino antisense oligonucleotide injection was used to knock down spatacsin function in zebrafish embryos. Examination of spg11 morphant embryos revealed a range of developmental defects and CNS abnormalities, and analysis of axon pathway formation demonstrated an overall perturbation of neuronal differentiation. These data confirm loss of spatacsin as the cause of SPG11-linked HSP-TCC in Asian kindreds, expanding the mutation spectrum recognized in this disorder. This study represents the first investigation in zebrafish addressing the function of a causative gene in autosomal recessive HSP and identifies a critical role for spatacsin during early neural development in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) encompasses a group of inherited neurological disorders characterized by degeneration of the corticospinal tract motor neurons and resulting in progressive lower limb spasticity. Over 35 HSP (or SPG) loci have been reported [1] and 18 disease genes identified, accounting for autosomal dominant [2–10], autosomal recessive [11–17], and X-linked [18, 19] subtypes of HSP. Importantly, many of the protein products of these genes share a role in intracellular transport and axonal trafficking [1, 20], suggesting that a common pathogenic mechanism may underlie a number of these disorders.
Hereditary spastic paraplegia with thin corpus callosum (HSP-TCC) is a distinct “complicated” form of autosomal recessive (AR) HSP associated with thinning of the corpus callosum. Despite phenotypic overlap with other ARHSPs, including Mast syndrome (SPG21) [14], agenesis of the corpus callosum with peripheral neuropathy (ACCPN) [21], and a form of HSP-TCC with epilepsy [22], the majority of HSP-TCC cases appear to be linked to the SPG11 locus on chromosome 15q15.1-q21.1 [23–27]. The SPG11 gene has been cloned and mutations identified in 11 of 12 European and North African families analyzed [15]. Since then, independent confirmation that spatacsin loss-of-function mutations cause HSP-TCC has been reported in patients of European, North African, Turkish, Middle Eastern, South American, and Asian origin. In this paper, we present further verification of the SPG11 gene as a major genetic determinant in HSP-TCC by describing novel mutations in four kindreds of Asian descent.
To gain further insight into the physiological and cellular bases of HSP-TCC pathology, we have also used morpholino-based gene knockdown in the zebrafish, Danio rerio, to investigate spg11 function during development. Heterozygous mutations in the human SPG4 and SPG8 genes, encoding spastin and strumpellin, respectively, cause autosomal dominant HSP with predominantly adult onset of disease [2, 10]. Previous investigations have shown that knockdown of spg4 and spg8 during zebrafish development is characterized by aberrant motor neuron axon outgrowth and widespread CNS developmental defects [10, 28]. Here, we report initial studies to address the function of a gene underlying autosomal recessive HSP.
Materials and methods
Patients
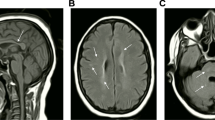
We assessed six affected and seven unaffected family members from four HSP-TCC kindreds of Indian (families 1, 3, and 4) and Pakistani (family 2) descent. Parental consanguinity was documented in all families. The diagnosis of HSP-TCC was established according to published criteria [22]. On examination, all affected individuals demonstrated significant lower limb spasticity, brisk deep tendon reflexes, and bilateral extensor plantar responses. Onset of symptoms occurred between 16 and 20 years of age, with the exception of the two affected siblings in family 3 presenting with subnormal intelligence and difficulty in walking since preschool age. Affected female probands from families 2 and 4 also had mild learning difficulties. Brain MRI revealed substantial thinning and hypoplasia of the corpus callosum in all affected cases, together with generalized moderate cerebral atrophy.
The study was approved by the University Hospitals of Leicester and Guy’s & St Thomas’ NHS Foundation Trust local research ethics committees, and all participants provided written informed consent. Control samples were of mixed Asian origin, ascertained in relation to a program of autozygosity mapping with inbred kindreds [29].
Genetic analysis
DNA was extracted from peripheral blood leukocytes by standard techniques. Microsatellite markers flanking the ACCPN, SPG11, and SPG21 loci were identified from the NCBI UniSTS database and amplified by PCR. Genotypes were assigned from PCR products using an ABI3730xl automated sequencer and GeneMapper v3.7 software (Applied Biosystems, Foster City, CA, USA). To confirm identity-by-descent, additional polymorphic CA-repeat regions were detected at 43.416 and 43.775 Mb and genotyped as above (primer details available on request). Marker positions were assigned according to the NCBI sequence map (Build 37.1).
All coding exons and intron–exon boundaries of the SPG21 and SPG11 genes were screened by direct DNA sequencing. Primers were designed using Primer3 software [30] or were as previously described [15]. PCR products were purified with ExoSAP-IT (GE Healthcare, Munich, Germany), sequenced using BigDye Terminator v3.1 chemistry (Applied Biosystems), and analyzed on an ABI3730xl automated sequencer. Sequence traces were aligned to reference using Sequencher v4.5 software (Gene Codes Corporation, Ann Arbor, MI, USA).
To directly assess the consequence of the intron 2 splice site mutation upon transcript translation, total RNA was extracted from peripheral blood of subjects III:2 and IV:3 with the QIAamp RNA blood mini kit (Qiagen, Crawley, UK). First-strand complementary DNA (cDNA) synthesis was performed using the Reverse-iT 1st strand synthesis kit (ABgene, Epsom, UK). Gene-specific primers SPG11ex1intF (5′ CAGCCTCCAAGTGCTTTCTT 3′) and SPG11ex4intR (5′ GCTCCTGTTGCTGCTCATTA 3′) were used for reverse transcriptase PCR (RT-PCR) and sequencing of messenger RNA (mRNA).
Zebrafish maintenance
Zebrafish embryos were obtained from natural matings, maintained at 28.5°C, and staged as previously described [31]. Pigmentation was inhibited by treatment with 0.2 nM phenylthiourea at 24 h post-fertilization (hpf).
Bioinformatics and cloning of the zebrafish ortholog
Searching the zebrafish genome assembly (Zv7 release v48) for KIAA1840 orthologs identified an Ensembl gene (ENSDARG00000045968) on chromosome 25, within contig Zv7_scaffold2404.1, at position 317,067–343,586 kb. This gene encodes several overlapping predicted transcripts sharing homology with the 3′ region of human SPG11 cDNA (exons 25–40). To delineate the rest of the zebrafish gene, human cDNA sequence was queried against the zebrafish genome and EST databases using the TBLASTN algorithm. A predicted partial mRNA (XM_001346277), located within Zv7_NA7051.1-2, was identified exhibiting homology with human SPG11 exons 2–9. Using primer pairs JH37 (5′ TTCTTGTGGGAGGATGTGAG 3′) and JH42 (5′ CTCCGCTCAGCAGGACTCT 3′) and JH43 (5′ GCAGGTGAGCGTCTGATTTT 3′) and JH44 (5′ AGGAAGCTGTTGGTCTTGGA 3′), we generated partial cDNA fragments of 1,522 and 1,097 bp. Both PCR products were cloned into the pCRII-TOPO vector (Invitrogen, Paisley, UK) to create constructs pCR3′spg11 and pCR5′spg11. The central 4,575-bp portion (exons 9–33) was TA-cloned into the pGem-T Easy vector (Promega, Madison, WI, USA), by long-range RT-PCR with primers zSPG11ex7intF (5′ GTTCTTCATCCGGCTTCAGT 3′) and zSPG11ex34intR (5′ CGAACAATCCCTTCCAGATT 3′). All clones were verified using internal sequencing primers. Multiple sequence alignments were generated using MultAlin software [32], and motif analysis was performed with QuasiMotiFinder [33].
5′ Ready amplification of cDNA ends
To determine exon 1 of the zebrafish spg11 gene, 5′ ready amplification of cDNA ends (5′ RACE) was performed using the GeneRacer RACE ready cDNA kit (Invitrogen) according to the manufacturer’s guidelines. The gene-specific reverse primer JH40b (5′ AACACACACGTGTTCACCAACAACAC 3′) was used for RT-PCR amplification with the forward GeneRacer 5′ Primer, and a nested PCR was performed using zSPG11ex2-3intR (5′ ATGTCGGCGTGTGTGTGTTTGATGA 3′) and GeneRacer 5′ Nested Primer. PCR products were gel excised and sequenced directly with primer zSPG11ex2-3intR. The ATG start site was identified by translating all three reading frames of the mRNA sequence using publicly accessible software [34] and correlating zebrafish and human sequences.
Whole-mount in situ hybridization and immunohistochemistry
In situ hybridization was performed as previously described [35]. Antisense and sense digoxigenin-labeled RNA probes were generated by in vitro transcription from plasmids pCR5′spg11 and pCR3′spg11. To address the role of spg11 during CNS development, axon pathway formation was analyzed by anti-acetylated α-tubulin immunohistochemical labeling at 52 hpf. Alkaline phosphate conjugated antibodies (6-11B-1) were used for detection (Roche Applied Science, Mannheim, Germany and Sigma-Aldrich, St. Louis, MO, USA). Embryos were mounted in 90% glycerol and photographed using a Nikon DS-5Mc cooled camera system with NIS-Elements Basic Research (BR) v3.0 software (Nikon Instruments Inc., Amstelveen, The Netherlands).
Morpholino knockdown of zebrafish spg11
The antisense splice-blocking morpholino oligonucleotides spg11E2I2 (5′ GTGATGTTCAGTTCTCTTACCGGCG 3′) and spg11E4I4 (5′ TGTTGGCGGTGTCTGTGCACCTGAA 3′), designed to target the spg11 exon2–intron2 and exon4–intron4 boundaries, respectively, were synthesized by Gene Tools, LLC (Philomath, OR, USA). The 5-mismatch morpholino spg11E2I2mm (5′ GTcATcTTgAGTTCTCTTAgCcGCG 3′) was used as a specificity control. Lyophilized oligonucleotide resuspended in sterile water was used at a working concentration of 2 ng/µl containing 0.1% phenol red and rhodamine for visualization and sorting of injected embryos. Titrations were performed by injecting 0.5–2 ng into a single blastomere of 1–2 cell stage embryos, and an optimal dose minimizing mortality and toxic effects was determined. Total RNA was isolated from embryos to detect aberrant splicing events using RT-PCR with primers JH49 (5′ TCTGAGCAGGAGTGATGCTG 3′) and JH40 (5′ AACACACACGTGTTCACCAAC 3′) across exon 2 and zSPG11ex3intF (5′ CGTCTGTGAGGCTGTTGTGT 3′) and zSPG11ex6intR (5′ TCTCGTCTGTCTGCGCTCTA 3′) across exon 4. PCR products were isolated by gel purification and sequenced.
Results
Genotyping and mutation detection
Microsatellite genotyping in families 1 and 2 revealed all affected subjects were heterozygous for ACCPN markers, but showed homozygosity at the SPG11 (Fig. 1) and SPG21 loci (data not shown). Sequencing of SPG21 excluded this gene as the cause of HSP-TCC in both families by the identification of heterozygous SNPs in affected subjects. Genotyping data for families 3 and 4 were consistent with exclusion of the ACCPN and SPG21 loci, having heterozygous genotypes in affected cases, and were homozygous for SPG11 (Fig. 1). Following additional microsatellite genotyping across the SPG11 locus, the minimal disease gene interval was defined to a 3.96-Mb region, flanked by markers D15S779 and D15S659 (Figs. 1 and 2a).

Pedigree structures of the four families with genotyping data across the SPG11 locus. Pedigree identifiers indicate subjects from which DNA was available. All affected subjects are homozygous for microsatellite markers across this region. Critical recombinations in families 1 and 3 define the disease gene interval between markers D15S779 and D15S659

Novel SPG11 mutations in HSP-TCC families. a Physical map of chromosome 15q15.1-q21.3, showing the 3.96-Mb minimal interval harboring the SPG11 (KIAA1840) gene. b Schematic of the SPG11 gene indicating the locations of the identified mutations. c Sequence chromatograms. Family 1 carries a c.422+1G>A substitution at the first nucleotide of intron 2, indicated by the asterisk. Affected subjects from family 3 carry a c.2146C>T (p.Q716X) nonsense mutation in exon 11. Amino acid codes are depicted above the sequence traces. A novel c.3602_3603delAT (p.Y1201LfsX4) frameshift mutation in exon 21 was detected in family 4. The proband in family 2 has a c.4846C > T (p.Q1616X) nonsense mutation in exon 28. All unaffected parents are heterozygous for identified mutations. d RT-PCR analysis in individuals Fam.1-III:2 and -IV:3 shows a wild-type fragment of 575 bp and mutant fragments of 390 bp. Direct sequencing of the mutant band confirms skipping of exon 2 with contiguous sequence from the end of exon 1 into the start of exon 3
Sequence analysis of the SPG11 gene revealed a splice donor site mutation in intron 2 (c.442+1G>A), co-segregating with the disease phenotype in family 1 (Fig. 2b, c). Affected subjects were homozygous and all unaffected family members were heterozygous for this mutation, consistent with the genotyping data generated previously. Using primers in exons 1 and 4, RT-PCR in a heterozygous parent and an affected subject revealed a wild-type fragment of 575 bp and a smaller mutant band of 390 bp, the expected size upon excision of exon 2. Sequencing of the mRNA transcript confirmed skipping of exon 2 in the mutant allele, resulting in premature termination and a truncated protein (p.H86QfsX15; Fig. 2d).
Mutation screening of the SPG11 gene in the remaining HSP-TCC kindreds identified three further novel mutations (Fig. 2c). The sequence variants detected in families 2 and 3 also predict generation of truncated transcripts due to homozygous c.4846C>T and c.2146C>T nonsense mutations in exons 28 and 11, incorporating stop codons in place of the native glutamines at positions 1616 and 716, respectively. In family 4, a homozygous 2-bp deletion was detected in exon 21 (c.3602_3603delAT), predicting a frameshift leading to premature termination of the polypeptide chain four amino acids downstream (p.Y1201LfsX4). None of the identified mutations were observed in a panel of 180 control chromosomes of similar ethnicity.
Isolation and characterization of the zebrafish spg11 gene
Reference sequence for ∼1.5 kb at each terminus of the spg11 ortholog was obtained through bioinformatics and 5′ RACE, facilitating amplification of the central portion by RT-PCR. Complete transcript sequence data have been deposited with the GenBank data library under accession numbers FJ217177 (full-length cDNA, 7,140 bp) and FJ217178 (alternative cDNA, 3,747 bp). Evaluation of sequence homology between human and zebrafish spatacsin revealed 41% identity and 60% similarity across the length of the proteins (Fig. 3), with particularly high levels of nucleotide sequence identity across human SPG11 exons 35–36. Using a multiple sequence alignment across vertebrate species (data not shown), a highly conserved putative WD repeat signature within this region was subsequently detected with QuasiMotiFinder (Fig. 3).

Multiple sequence alignment of human and zebrafish spatacsin proteins. Human reference sequence was obtained from the Ensembl database (ENSP00000261866). Identical amino acid residues are highlighted in dark gray and residues sharing more than 90% similarity in light gray. Putative functional domains (i–iv), indicated by horizontal lines, correspond to the glycosyl hydrolase family 1, leucine zipper, coiled coil, and Myb domains described, previously [15]. The highly conserved WD repeat signature predicted by QuasiMotiFinder is boxed (Prosite ref. PS00678)
Expression of spg11 during zebrafish development
Developmental RT-PCR analysis revealed that spg11 was expressed both maternally and zygotically, persisting until at least day 3 of development. Using whole-mount in situ hybridization, spg11 transcripts were ubiquitously distributed throughout the embryo at 26 hpf, with increased expression levels detected in the brain and low levels in the notochord. Corresponding sense probes produced no significant hybridization signal (data not shown).
Morpholino knockdown and immunohistochemistry
RT-PCR analysis of spg11E2I2 morphant embryos detected three transcripts: the wild-type 468-bp fragment, an abundant 413-bp product, corresponding to an mRNA lacking the terminal 55 bp of exon 2 and a less abundant 551-bp fragment caused by retention of intron 2. Both mis-splicing events are predicted to severely truncate the encoded proteins through introduction of a premature stop codon in exon 3 (Fig. 4a), providing a good model of the donor splice site mutation detected in family 1. Morpholino titration to determine an effective dose for phenotypic analysis revealed a dose-dependent blocking of splicing (Fig. 4b). Embryos injected with 0.5 ng demonstrated phenotypic recovery by 52 hpf, while 2 ng was toxic. The optimal dose for further analysis of spg11 morphants was therefore established at 1 ng.

Knockdown of zebrafish spg11 function using a splice blocking morpholino. a Representation of the mRNA transcripts detected in spg11 control and morphant embryos. Intron–exon boundaries are depicted in the genomic schematic across exons 1–3. The asterisk denotes the ATG translational start site. Wild-type mRNA produces a 468-bp PCR product with primers JH49 and JH40. Alternatively spliced transcripts from morphant embryos (E2I2a and E2I2b) lead to premature truncation of translated products by incorporation of a stop codon (TGA) in exon 3. b Gel image showing RT-PCR products detected in 26 and 52 hpf zebrafish across a titration of morpholino doses. Wild-type (468 bp) and alternatively spliced (413 and 551 bp) PCR products demonstrate that splice blocking is dose-dependent. c–g Lateral view of the brain. At 26 hpf, morphant embryos have defective brain ventricle formation (d, arrow) and smaller eyes. At 52 hpf, spg11 morphant brains have mild hydrocephaly and a reduced mid-hindbrain boundary (mhb, arrow) (g). h, i Lateral view of the trunk at 52 hpf. The typical chevron somite pattern (dashed lines) is flatter in spg11 morphants. j–s Anti-acetylated α-tubulin labeling of control (j, l, n, p, r) and spatacsin morphant embryos (k, m, o, q, s) at 52 hpf. Embryos were injected with 1 ng of spg11E2I2 morpholino. j, k Ventral view of the brain showing formation of the optic axons (oa) with reduced retinal ganglion cell differentiation in spg11 morphants. l, m Lateral view of the brain highlighting reduced differentiation of facial motor (VII) and branchiomotor (X) neurons in morphant embryos. n–q Dorsal views of the forebrain, midbrain, and hindbrain. The posterior commissure (pc) forms but the habenular commissure (hc) fails to project to the dorsal midline (o, arrow). Morphant hindbrains demonstrate a severe reduction in neuronal differentiation with an absence of glial curtain cells and formation of an ectopic cluster of disorganized neurons (q, arrow). r, s Lateral view of the trunk showing impaired projection and orientation of the spinal motor neuron axons. ac anterior commissure, hb hindbrain, tec tectum, tel telencephalon
A range of developmental defects was apparent in spg11E2I2 morphants by 26 hpf. Morphologically, the embryos had a curly-tail down phenotype (data not shown), defective brain ventricle formation, and small eyes (Fig. 4c–e). At 52 hpf, spg11 morphants had developed mild hydrocephaly, the mid-hindbrain boundary was reduced, and the eyes were still smaller (Fig. 4f, g). In the trunk, the typical chevron somite pattern appeared flatter in morphant embryos in comparison to uninjected controls (8% mild, 36% moderate, 55% severely affected embryos at 1-ng morpholino dose; Fig. 4h, i).
Morphant embryos exhibited a generalized perturbation of neuronal differentiation, with reduced levels of staining visible in spg11 morphants compared to controls (Fig. 4j–s). Although retinal ganglion cell differentiation was reduced in the eye, the optic axons did form and exit the retina correctly before projecting to the tectum (Fig. 4j, k). Analysis of cranial motor neurons revealed that the facial and branchiomotor neurons of spg11 morphant embryos were present, but appeared shorter and less differentiated than those of control embryos (Fig. 4l, m). The posterior commissure also formed normally by 52 hpf; however, the habenular commissure failed to project to the dorsal midline (Fig. 4n, o). In the hindbrain, neuronal differentiation was severely reduced and glial “curtain” cells were absent. In addition, an ectopic cluster of disorganized neurons was often apparent within spg11 morphant hindbrains (Fig. 4p, q). In the trunk, spinal motor neuron axons projected correctly but fascicules appeared thinner, suggesting a reduced number of neurons. Furthermore, there was some evidence of impaired orientation and axons appeared shortened, implying a failure to reach their ventral target (Fig. 4r, s). Embryos injected with an equivalent dose of the spg11E2I2 mismatch oligonucleotide appeared comparable to uninjected controls, indicating that the observed phenotype is due to knockdown of the spg11 transcript (Electronic supplementary materials).
To confirm the specificity of reduction of spg11 activity, zebrafish embryos were injected with a second spg11-specific morpholino targeted to the intron 4 splice donor site, mutation of which has also been previously reported in the human SPG11 gene [36]. RT-PCR analysis of embryos injected with the spg11E4I4 morpholino demonstrated efficient splice blocking, detecting an alternatively spliced transcript corresponding to retention of intron 4.
Marked CNS abnormalities were also apparent in spg11E4I4 morphants, and in comparison to spg11E2I2-injected embryos, a more severe truncation of the spinal motor neuron axons was observed at higher concentrations (Electronic supplementary materials). In addition to the CNS features, embryos also exhibited a range of developmental defects, including an enlarged heart cavity, which is a relatively common feature in injected fish, a curly tail and deformities of the fin, and were generally smaller than the uninjected controls. As observed with the spg11E2I2 morpholino, the maximum dose tested was toxic. Statistical analysis of the distribution of morphological abnormalities by one-way ANOVA revealed a significant difference between target and control morpholinos at the 1-ng dose (p < 0.0001; Table 1).
Discussion
The genetic basis of the hereditary spastic paraplegias is rapidly evolving with 18 disease genes now reported, accounting for approximately 50% of the known loci. In this study, we report the genetic analysis of four autosomal recessive HSP-TCC families, each originating from the Indian subcontinent. Following localization to chromosome 15 by autozygosity mapping, direct sequencing of the SPG11 gene identified novel pathogenic homozygous mutations (c.442+1G>A, c.2146C>T, c.3602_3603delAT, and c.4846C>T) in all kindreds. To date, spatacsin mutations have predominantly been reported in European (47%), North African (14%), and Chinese (11%) populations, as detailed in Electronic supplementary materials. However, only one study has described SPG11 mutations in patients of subcontinental descent [37], most likely representing an ascertainment bias but suggesting that SPG11 variation may be a rare cause of HSP in these patients.
All mutations identified in our patients lead to a truncated protein product, consistent with other reports that loss of spatacsin function is the pathogenic mechanism underlying SPG11-linked HSP. While a homozygous G>C substitution has previously been reported at the intron 2 splice donor site [38], the variant segregating in family 1 is the first example of a c.442+1G>A mutation and formally demonstrates that aberrant splicing results in exon 2 skipping. Families 2 and 4 harbor mutations in exons 28 and 21 of the SPG11 gene. Variants have previously been identified in more than half of all exons; however, <10% of HSP-TCC cases are accounted for by mutations in exons 18–29, encoding the central region of the spatacsin protein. Interestingly, previous bioinformatic analysis has indicated this to be a region of putative structural importance [15]. Although the function of spatacsin remains undetermined, the putative leucine zipper motif and Myb domain have implied a possible regulatory function during gene expression. Through resolution of sequence and amino acid homology between human and zebrafish, we detected a highly conserved WD repeat signature across vertebrate orthologs of spatacsin. Characteristically, WD proteins are composed of repeating units culminating in tryptophan (W) and aspartic acid (D) and share a functional role in the regulation of cellular processes such as cell division, gene transcription, transmembrane signaling, mRNA modification, and vesicle fusion [39]. It remains to be determined whether the restricted motif detected here reflects a similar function for spatacsin.
To investigate the role of spatacsin during development, we have performed whole-mount in situ hybridization studies in the zebrafish embryo. Previous studies of Spg11 mRNA expression in the rat brain have demonstrated that it is undetectable in the newborn but is present in the cerebellum from postnatal days 6–21 [15]. In contrast, RT-PCR in the zebrafish embryo has detected spg11 mRNA expression both maternally and zygotically, persisting until at least 72 hpf. Whole-mount in situ hybridization revealed that transcripts were ubiquitously distributed throughout the embryo at 26 hpf with high levels of expression in the CNS. This ubiquitous expression, similar to that reported for spg4 [28], is consistent with previous RT-PCR profiling of the SPG11 gene in human tissues [40] and suggests an important role for spatacsin during early developmental processes throughout the embryo.
To specifically assess the functional impact of the c.442+1G>A splice site mutation, we sought to recapitulate the phenotype in zebrafish by means of morpholino injection. We observed a range of developmental defects in spg11 morphant embryos, including CNS abnormalities. Overall, spg11 morphants exhibited perturbed neuronal differentiation, with markedly reduced levels of immunohistochemical staining in comparison to control embryos. Consistent with the features seen in spg4 and spg8 embryos [10, 28], spg11 morphants had a curly-tail phenotype and motor neuron axons appeared reduced with some evidence of impaired orientation, albeit less pronounced than in the autosomal dominant HSP counterparts. While axonal structures within the brain developed normally, subsequent neuronal projections appeared to be impeded, particularly within the forebrain and midbrain regions. The characteristic lower limb spasticity in HSP is caused by degeneration of the corticospinal tract motor neurons. The data presented in this study are in agreement with previous reports demonstrating a disruption of axon morphogenesis in loss of HSP-specific gene activity [10, 28], and while the function of the strumpellin protein remains unknown, this may suggest a similar role for spatacsin in microtubule dynamics. Further studies will be required to determine the neuronal-specific effects of spg11 expression during later development. In humans, autosomal recessive HSP-TCC typically manifests during infancy or puberty, and slow disease progression continues over a period of 10–20 years. In addition to lower limb spasticity and cognitive impairment, some individuals exhibit additional features including dysarthria, dysphagia, and ocular defects. Interestingly, spatacsin knockdown also appears to impair eye development in spg11 morphants.
In summary, this study provides confirmation that loss of spatacsin function is the primary molecular defect in the pathogenesis of SPG11-linked HSP-TCC and points to a critical role for spatacsin during early neural development. For clarity, we have also collated all SPG11 mutations reported to date, providing a comprehensive resource for neurologists and molecular geneticists in genetic screening for HSP-TCC.
References
Contino G, Novelli G (2006) Hereditary spastic paraplegia: clinical genomics and pharmacogenetic perspectives. Expert Opin Pharmacother 7:1849–1856
Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Dürr A, Wincker P, Brottier P, Cattolico L, Barbe V, Burgunder JM, Prud'homme JF, Brice A, Fontaine B, Heilig B, Weissenbach J (1999) Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 23:296–303
Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L, Bui M, Fink JK (2001) Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet 29:326–331
Hansen JJ, Dürr A, Cournu-Rebeix I, Georgopoulos C, Ang D, Nielsen MN, Davoine CS, Brice A, Fontaine B, Gregersen N, Bross P (2002) Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet 70:1328–1332
Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA (2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71:1189–1194
Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK (2003) NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 73:967–971
Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Hörl G, Malli R, Reed JA, Dierick I, Verpoorten N, Warner TT, Proukakis C, Van den Bergh P, Verellen C, Van Maldergem L, Merlini L, De Jonghe P, Timmerman V, Crosby AH, Wagner K (2004) Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet 36:271–276
Mannan AU, Krawen P, Sauter SM, Boehm J, Chronowska A, Paulus W, Neesen J, Engel W (2006) ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am J Hum Genet 79:351–357
Züchner S, Wang G, Tran-Viet KN, Nance MA, Gaskell PC, Vance JM, Ashley-Koch AE, Pericak-Vance MA (2006) Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet 79:365–369
Valdmanis PN, Meijer IA, Reynolds A, Lei A, MacLeod P, Schlesinger D, Zatz M, Reid E, Dion PA, Drapeau P, Rouleau GA (2007) Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet 80:152–161
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93:973–983
Eymard-Pierre E, Lesca G, Dollet S, Santorelli FM, di Capua M, Bertini E, Boespflug-Tanguy O (2002) Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am J Hum Genet 71:518–527
Patel H, Cross H, Proukakis C, Hershberger R, Bork P, Ciccarelli FD, Patton MA, McKusick VA, Crosby AH (2002) SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet 31:347–348
Simpson MA, Cross H, Proukakis C, Pryde A, Hershberger R, Chatonnet A, Patton MA, Crosby AH (2003) Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet 73:1147–1156
Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, Martin E, Ouvrard-Hernandez AM, Tessa A, Bouslam N, Lossos A, Charles P, Loureiro JL, Elleuch N, Confavreux C, Cruz VT, Ruberg M, Leguern E, Grid D, Tazir M, Fontaine B, Filla A, Bertini E, Durr A, Brice A (2007) Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 39:366–372
Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, Benomar A, Lossos A, Denora P, Fernandez J, Elleuch N, Forlani S, Durr A, Feki I, Hutchinson M, Santorelli FM, Mhiri C, Brice A, Stevanin G (2008) Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 82:992–1002
Tsaousidou MK, Ouahchi K, Warner TT, Yang Y, Simpson MA, Laing NG, Wilkinson PA, Madrid RE, Patel H, Hentati F, Patton MA, Hentati A, Lamont PJ, Siddique T, Crosby AH (2008) Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am J Hum Genet 82:510–515
Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson J, Metzenberg A, Ionasescu V, Temple K, Kenwrick S (1994) X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet 7:402–407
Saugier-Veber P, Munnich A, Bonneau D, Rozet JM, Le Merrer M, Gil R, Boespflug-Tanguy O (1994) X-linked spastic paraplegia and Pelizaeus–Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet 6:257–262
Soderblom C, Blackstone C (2006) Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther 109:42–56
Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière JB, Prévost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA (2002) The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32:384–392
Al-Yahyaee S, Al-Gazali LI, De Jonghe P, Al-Barwany H, Al-Kindi M, De Vriendt E, Chand P, Koul R, Jacob PC, Gururaj A, Sztriha L, Parrado A, Van Broeckhoven C, Bayoumi RA (2006) A novel locus for hereditary spastic paraplegia with thin corpus callosum and epilepsy. Neurology 66:1230–1234
Shibasaki Y, Tanaka H, Iwabuchi K, Kawasaki S, Kondo H, Uekawa K, Ueda M, Kamiya T, Katayama Y, Nakamura A, Takashima H, Nakagawa M, Masuda M, Utsumi H, Nakamuro T, Tada K, Kurohara K, Inoue K, Koike F, Sakai T, Tsuji S, Kobayashi H (2000) Linkage of autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum to chromosome 15q13-15. Ann Neurol 48:108–112
Casali C, Valente EM, Bertini E, Montagna G, Criscuolo C, De Michele G, Villanova M, Damiano M, Pierallini A, Brancati F, Scarano V, Tessa A, Cricchi F, Grieco GS, Muglia M, Carella M, Martini B, Rossi A, Amabile GA, Nappi G, Filla A, Dallapiccola B, Santorelli FM (2004) Clinical and genetic studies in hereditary spastic paraplegia with thin corpus callosum. Neurology 62:262–268
Lossos A, Stevanin G, Meiner V, Argov Z, Bouslam N, Newman JP, Gomori JM, Klebe S, Lerer I, Elleuch N, Silverstein S, Durr A, Abramsky O, Ben-Nariah Z, Brice A (2006) Hereditary spastic paraplegia with thin corpus callosum: reduction of the SPG11 interval and evidence for further genetic heterogeneity. Arch Neurol 63:756–760
Ölmez A, Uyanik G, Özgül RK, Gross C, Cirak S, Elibol B, Anlar B, Winner B, Hehr U, Topaloglu H, Winkler J (2006) Further clinical and genetic characterization of SPG11: hereditary spastic paraplegia with thin corpus callosum. Neuropediatrics 37:59–66
Stevanin G, Montagna G, Azzedine H, Valente EM, Durr A, Scarano V, Bouslam N, Cassandrini D, Denora PS, Criscuolo C, Belarbi S, Orlacchio A, Jonveaux P, Silvestri G, Hernandez AM, De Michele G, Tazir M, Mariotti C, Brockmann K, Malandrini A, van der Knapp MS, Neri M, Tonekaboni H, Melone MA, Tessa A, Dotti MT, Tosetti M, Pauri F, Federico A, Casali C, Cruz VT, Loureiro JL, Zara F, Forlani S, Bertini E, Coutinho P, Filla A, Brice A, Santorelli FM (2006) Spastic paraplegia with thin corpus callosum: description of 20 new families, refinement of the SPG11 locus, candidate gene analysis and evidence of genetic heterogeneity. Neurogenetics 7:149–156
Wood JD, Landers JA, Bingley M, McDermott CJ, Thomas-McArthur V, Gleadall LJ, Shaw PJ, Cunliffe VT (2006) The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum Mol Genet 15:2763–2771
Baumber L, Tufarelli C, Patel S, King P, Johnson CA, Maher ER, Trembath RC (2005) Identification of a novel mutation disrupting the DNA binding activity of GCM2 in autosomal recessive familial isolated hypoparathyroidism. J Med Genet 42:443–448
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana, Totowa, pp 365–386
Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203:253–310
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890
Gutman R, Berezin C, Wollman R, Rosenberg Y, Ben-Tal N (2005) QuasiMotiFinder: protein annotation by searching for evolutionarily conserved motif-like patterns. Nucleic Acids Res 33:W255–W261
Bikandi J, San Millán R, Rementeria A, Garaizar J (2004) In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics 20:798–799
Jowett T, Lettice L (1994) Whole-mount in situ hybridizations on zebrafish embryos using a mixture of digoxigenin- and fluorescein-labelled probes. Trends Genet 10:73–74
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, Rosa AL, Lerer I, Hamri A, Alegria P, Loureiro J, Tada M, Hannequin D, Anheim M, Goizet C, Gonzalez-Martinez V, Le Ber I, Forlani S, Iwabuchi K, Meiner V, Uyanik G, Erichsen AK, Feki I, Pasquier F, Belarbi S, Cruz VT, Depienne C, Truchetto J, Garrigues G, Tallaksen C, Tranchant C, Nishizawa M, Vale J, Coutinho P, Santorelli FM, Mhiri C, Brice A, Durr A, SPATAX consortium (2008) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131:772–784
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70:1384–1389
Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W, Uyanik G, Engel A, Lenz D, Seibel A, Hehr A, Ploetz S, Gamez J, Rolfs A, Weis J, Ringer TM, Bonin M, Schuierer G, Marienhagen J, Bogdahn U, Weber BH, Topaloglu H, Schols L, Riess O, Winkler J (2007) Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 62:656–665
Neer EJ, Schmidt CJ, Nambudripad R, Smith TF (1994) The ancient regulatory-protein family of WD-repeat proteins. Nature 371:297–300
Nagase T, Nakayama M, Nakajima D, Kikuno R, Ohara O (2001) Prediction of the coding sequences of unidentified human genes. XX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 8:85–95
Acknowledgments
This work was supported by the Wellcome Trust (programme grant 062346/Z/00/Z, project grant 078751/Z/05/Z). The authors would like to express their gratitude to the families for participating in this study and to Shanta Patel, University Hospitals of Leicester NHS Trust, for facilitating sample collection. We also acknowledge the assistance provided in extracting, storing, and forwarding DNA samples for genetic analysis by the Human Brain Tissue Repository DNA Bank (Human Brain Bank), Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore.
Ethical standards
These experiments comply with the current laws of the United Kingdom.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Southgate, L., Dafou, D., Hoyle, J. et al. Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 11, 379–389 (2010). https://doi.org/10.1007/s10048-010-0243-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-010-0243-8