
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare congenital disease with autosomal dominant transmission characterized by the presence of malformations of the big toes and of postnatal progressive heterotopic endochondral osteogenesis. We report the case of 3-year-old girl with dysplasia of the first metatarsal bones and progressive heterotopic ossificans of the right thigh due to previous diphtheria–tetanus–pertussis immunizations and several inappropriate surgical interventions. Direct sequence analysis identified a 617G–A nucleotide mutation in the patient but not in her parents or brother. Pedigree analysis suggests that a de novo mutation in the ACVR1 gene is responsible for the disease in this family. This is the first report of the results of a mutation analysis in a sporadic case of FOP in a Taiwanese patient.
Similar content being viewed by others

Introduction
Fibrodysplasia ossificans progressiva (FOP) is a rare congenital disease with autosomal dominant transmission (Kaplan et al. 1993a), characterized by the presence of malformations of the big toes and of postnatal progressive heterotopic endochondral osteogenesis of tendons, ligaments, fasciae and striated muscles, especially in the presence of exacerbating factors such as trauma, surgical intervention, lesion biopsy, and intramuscular injection (Cohen et al. 1993; Connor et al. 1993; Rocke et al. 1994; Lanchoney et al. 1995).
Recently, Shore et al. (2006) successfully mapped FOP to chromosome 2q23–24 by linkage analysis and identified an identical heterozygous mutation (617G –> A; R206H) in the glycine–serine (GS) activation domain of ACVR1, a BMP type I receptor, in all affected individuals examined.
We report the case of a 3-year-old girl with dysplasia of the first metatarsal bones and progressive heterotopic ossificans of the right thigh following diphtheria–tetanus–pertussis (DPT) immunizations in 2003 (Wang et al. 2003).
The aim of this study was to search for disease-associated mutations within the ACVR1 gene in our case. To our knowledge, this is the first genetic study of FOP in a Taiwanese patient.
Materials and methods
Patient
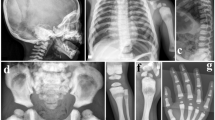
This 3-year-old girl was born by vaginal delivery at full-term. During her development, no abnormality was noted. Following the standard DPT immunization, two subcutaneous masses at the injection sites in her thigh were noted at the age of 6 and 18 months. Her parents brought her to visit a hospital due to restricted right hip motion that interfered with walking at the age of 2 years. She underwent surgical intervention twice for the right thigh lesion following a diagnosis of myositis ossificans. However, her right lower extremity disability did not improve and had even progressed by the time her parents brought her to our clinic at the age of 3 years. The gross appearance showed a palpable irregular hard mass over the right proximal thigh with the tip protruding under the lateral skin (Fig. 1a). The motion of the right hip and knee were restricted, with the hip stiff in ten degrees flexion and the knee range of motion being full extension to 20 degrees flexion with interference of diffusive heterotopic ossification apparent upon radiography (Fig. 1b). With the impression of post-traumatic myositis ossificans, we performed surgical excision of the ectopic bone to resolve the disability of the right hip and to obtain a sufficient amount of sample to confirm diagnosis (Fig. 1c). Initially the value of ALP decreased from 853 IU/l to 420 IU/l. There was dramatic improvement in the range of motion of the hip and knee. However, the symptoms flared up again 1 month after the operation, with ALP becoming elevated to 1,158 IU/L and a bony mass over the right thigh again developing. During the period of hospitalization, a bilateral first toe valgus deformity was noted and dysplasia of the bilateral first metatarsal bones was revealed by radiography (Fig. 1d). From a review of the literature, FOP was highly suspected. Other biochemical measurements, including thyroid function, serum calcium and others, were within normal range. However, the pathology study revealed a remarkable picture of fibrous dysplasia, with irregular woven bone and islands of cartilage (Fig. 1e). A follow-up plain film showed that heterotopic ossificans attacked again about 1 month later (Fig. 1f).

a Gross appearance of the right thigh. b Radiography of right femur revealing massive ectopic bone. c Ectopic bone exposed in the wound. d Radiography of patient’s feet, revealing dysplasia of both first metatarsal bones. e Photomicrography demonstrating irregularly shaped trabeculae of woven bone embedded in moderately cellular fibrous tissue. Osteoblasts are uniformly aligned along the edges of bony trabeculae, suggesting an intermediate stage of fibrodysplasia ossificans progressiva (FOP); (Hematoxylin and eosin, original magnification ×400.) f Radiographic study 1 month after surgical intervention, demonstrating a new episode of rapidly attacking heterotopic ossificans
Genetic analysis
This research was approved by the Kaohsiung Medical University Ethics Committee and all subjects participating in the study gave consent. Blood samples for the familial population were collected. Genomic DNA was isolated from peripheral blood leukocytes by the guanidine–HCl method. Amplification of each exon and promoter was performed individually by polymerase chain reaction (PCR). The oligonucleotide primers used in this study were: ACVR 1-f-TCAGGAAGTGGCTCTGGTCT, ACVR 1-r-CAAGTTCAGGTGCTCCAACA.
PCR was performed in a programmable DNA thermal cycler (Gene Amp PCR system 2400, Perkin-Elmer, Foster City, CA) for a total of 35 cycles under the following conditions: 95°C 30 s, 58°C 30 s and 72°C 30 s, with 95°C 5 min and 72°C 5 min for the first and the final cycle, respectively.
Direct DNA sequence analysis was performed by automated DNA sequencing in an ABI DNA sequencer (Model 377) with a dye terminator kit (Big Dye Terminator Cycle Sequencing V2.0). The results were further verified by restriction endonuclease digestion (Cac 8I).
Results
DNA sequence analysis demonstrated the presence of a single heterozygous nucleotide change at cDNA position 617 (617G–A) in our patient with FOP (Fig. 2b). The 617G–A nucleotide mutation was not found in any of her family members (Fig. 2d). In addition to direct DNA sequence analysis, we verified the G–A nucleotide change by restriction endonuclease digestion (Fig. 2b).

a Family pedigree. Filled symbols FOP-affected individuals. b DNA sequencing of the activin receptor type IA (ACVR1) gene, demonstrating the heterozygous mutation (R206H; 617G–A) in the patient with FOP. c The G–A mutation forms a new Cac8I site. Cac8I digestion of the PCR product from the G allele (lanes Father, Mother, Brother) generates two bands (196 bp and 48 bp), whereas the A allele (lane FOP) produces three bands (244, 196 and 48 bp). The samples from the individual with FOP is heterozygous for the two alleles. d wild type sequence was found in all other family members
Discussion
In 2006, Shore et al. identified an identical DNA sequence change (R206H) in the ACVR1 gene in every examined individual with FOP (Shore et al. 2006). The FOP R206H ACVR1 mutation is thus one of the most specific codons in the human genome to be associated with a disease phenotype, and was found in all populations analyzed, including African–American, European American, European (UK), Korean and Native Brazilian. Following this important report, here we add a Taiwanese patient with a de novo ACVR1 617G–A mutation.
Initially, from the evidence of two surgical interventions and elevation of ALP, we considered this patient as a case of post-traumatic myositis ossificans. We made a mistake in carrying out a surgical intervention in this case before correct diagnosis, and we wish to remind all orthopedic doctors and physicians to pay more attention to the feet of children with a history of heterotopic bone formation. Kaplan and most authors suggested that diagnosis of FOP is based on the presence of congenital malformation of the big toes and radiographic evidence of progressive heterotopic ossification in characteristic locations (Connor and Evans 1982; Kaplan et al. 1993a, b). Nowadays, the G–A nucleotide change distinguishing FOP from other disorders can be easily verified based on DNA sequence analysis and restriction endonuclease digestion, thus avoiding the mistake of further injury to the patient.
Reference
Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS (1993) The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am 75:215–219
Connor JM, Evans DA (1982) Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br 64:76–83
Connor JM, Skirton H, Lunt PW (1993) A three generation family with fibrodysplasia ossificans progressiva. J Med Genet 30:687–689
Kaplan FS, McCluskey W, Hahn G, Tabas JA, Muenke M, Zasloff MA (1993a) Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am 75:1214–1220
Kaplan FS, Tabas JA, Gannon FH, Finkel G, Hahn GV, Zasloff MA (1993b) The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J Bone Joint Surg Am 75:220–230
Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS (1995) Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatr 126:762–764
Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS (1994) Age- and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res 301:243–248
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Brown MA, Kaplan FS (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Wang HC, Huang PJ, Chen JC, Cheng YM (2003) A rare case report—fibrodysplasia ossificans progressiva. J Orthop Surg Taiwan 20:207–211
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Lin, GT., Chang, HW., Liu, CS. et al. De novo 617G–A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva. J Hum Genet 51, 1083–1086 (2006). https://doi.org/10.1007/s10038-006-0069-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0069-2
Keywords
This article is cited by
-
Phenotypic differences of patients with fibrodysplasia ossificans progressive due to p.Arg258Ser variants of ACVR1
Human Genome Variation (2015)
-
Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva
Clinical Rheumatology (2010)
-
Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements
European Journal of Human Genetics (2009)
-
A Recurrent Mutation c.617G>A in the ACVR1 Gene Causes Fibrodysplasia Ossificans Progressiva in Two Chinese Patients
Calcified Tissue International (2009)
-
The ACVR1 617G>A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva
Journal of Human Genetics (2007)
