
Abstract
Muscular dystrophies are a heterogeneous group of genetic disorders. In addition to genetic information, a combination of various approaches such as the use of genetic animal models, muscle cell biology, and biochemistry has contributed to improving the understanding of the molecular basis of muscular dystrophy's etiology. Several lines of evidence confirm that the structural linkage between the muscle extracellular matrix and the cytoskeleton is crucial to prevent the progression of muscular dystrophy. The dystrophin–glycoprotein complex links the extracellular matrix to the cytoskeleton, and mutations in the component of this complex cause Duchenne-type or limb-girdle-type muscular dystrophy. Mutations in laminin or collagen VI, muscle matrix proteins, are known to cause a congenital type of muscular dystrophy. Moreover, it is not only the primary genetic defects in the structural or matrix proteins, but also the primary mutations of enzymes involved in the protein glycosylation pathway that are now recognized to disrupt the matrix–cell interaction in a certain group of muscular dystrophies. This group of diseases is caused by the secondary functional defects of dystroglycan, a transmembrane matrix receptor. This review considers recent advances in understanding the molecular pathogenesis of muscular dystrophies that can be caused by the disruption of the cell–matrix linkage.
Similar content being viewed by others

Introduction
Muscular dystrophies are a heterogeneous group of genetic disorders characterized by the progressive loss of muscle strength and integrity. To date, a molecular genetic approach, such as positional cloning and candidate gene analysis, has identified more than 30 genes responsible for muscular dystrophies (Table 1) (for information on the location of mutations, see Stenson et al. 2003; http://www.hgmd.org). Clinically, these diseases present with muscle weakness at any age from birth to middle age, frequently with high serum creatine kinase levels, and result in significant morbidity and disability. At the level of muscle pathology, the muscular dystrophies are characterized by necrotic and regenerating fibers, an increase in fiber size variation, fiber splitting and centrally located myonuclei. Successive rounds of degeneration and regeneration of muscle fibers eventually result in necrosis and replacement of muscle with fatty and fibrous tissue. The dystrophic skeletal muscle plasma membranes are fragile, and membrane damage caused by muscle contraction leads to muscle cell wasting. The diseases have been classified by their clinical presentation, the distribution and extent of muscle weakness (some forms also affect the cardiac muscle), age of onset, rate of progression, and pattern of inheritance.
Genetic information has contributed to the understanding of the molecular pathogenesis of muscular dystrophy and to the development of rational strategies designed to alleviate the diseases. The first breakthrough in understanding the molecular basis of muscular dystrophy was the finding of the dystrophin gene, which is responsible for the common Duchenne muscular dystrophy (DMD) (Monaco et al. 1986; Hoffman et al. 1987). Subsequently, transmembrane proteins that form a large glycoprotein complex with dystrophin in the muscle plasma membrane, called the dystrophin–glycoprotein complex (DGC), were identified (Campbell and Kahl 1989; Yoshida and Ozawa 1990). One of the major functions of the DGC is to link the extracellular matrix to the intracellular dystrophin–cytoskeleton across the plasma membrane. Mutations in the components of the DGC can cause several forms of muscular dystrophy. Post-translational disruption of the matrix–cytoskeleton linkage due to mutations in enzymes is associated with congenital forms of muscular dystrophy. In addition, mutations in the components of the skeletal muscle basement membrane and the transmembrane matrix receptor proteins are known to cause muscular dystrophies. This genetic evidence indicates that muscle membrane integrity is maintained by the physical link between the extracellular matrix and the cytoskeleton. In this review, we will present the current understanding of the molecular pathogenesis of muscular dystrophies that are caused by primary genetic or secondary functional defects in matrix proteins, matrix receptors, and transmembrane proteins (Fig. 1; Table 1). We do not discuss diseases presumably caused by different pathomechanisms such as oculopharyngeal muscular dystrophy and myotonic dystrophy because several excellent reviews have been published (for example, see Morris 2000; Dalkilic and Kunkel 2003).

Schematic representation of the organization of molecules involving skeletal muscle cell–matrix linkage. Diagram illustrates the location of proteins involved in matrix–cell linkage. Genetic or functional disruption of this link causes muscular dystrophy. Basement membrane proteins laminin-2 and collagen VI are responsible for congenital muscular dystrophy 1A (classical type) and Ullrich/Bethlem diseases. Genetic or functional defects in transmembrane matrix receptor protein complexes cause several forms of muscular dystrophies, sarcoglycans for limb-girdle muscular dystrophies, α7 integrin for CMD, and DG for dystroglycanopathies. Dystrophin is responsible for Duchenne/Becker muscular dystrophies; ECM extracellular matrix; SG sarcoglycan; SSPN sarcospan; DG dystroglycan
DGC and muscular dystrophy
Duchenne muscular dystrophy is the most common form of muscular dystrophy, and the pattern of inheritance is X-linked recessive. The gene responsible for DMD was identified by using a positional cloning technique in the mid 1980s (Monaco et al. 1986; Hoffman et al. 1987). The gene product is dystrophin, which is a large cytoskeletal protein located at the cytoplasmic surface of the muscle plasma membrane (for a review of dystrophin, see Lapidos et al. 2004). The dystrophin gene is one of the largest genes known in humans and is composed of 79 exons. The size and complexity of the dystrophin gene may cause a high frequency of spontaneous mutation (approximately one third of DMD cases). Dystrophin is lost in the muscle of DMD patients, whereas it is reduced or truncated as a partially functional form in Becker muscular dystrophy, the milder alleic variant of DMD. The model mdx mice series has been used to understand the pathomechanism and develop therapeutic strategies: for example, virus-mediated gene transfer, exon skipping, and gene boost of utrophin (for a review, see van Deutekom and van Ommen 2003).
After the discovery of dystrophin, several transmembrane glycoproteins were identified (Campbell and Kahl 1989; Yoshida and Ozawa 1990). These proteins are co-purified with dystrophin and form a large protein complex that is called the dystrophin-glycoprotein complex (DGC) (Fig. 1). The components of the DGC include sarcoglycans (SG), sarcospan, and dystroglycan (DG) (for a review, see Michele and Campbell 2003). In dystrophin-deficient skeletal muscle, these components are concomitantly reduced from the muscle plasma membrane. It is believed that without the DGC, the muscle plasma membrane becomes fragile and susceptive to membrane damage produced by repeated cycles of muscle contraction and relaxation. DG is a transmembrane receptor of laminin, an extracellular matrix protein in the basement membrane, and intracellularly interacts with the C-terminal region of dystrophin. Dystrophin contains actin-binding domains at the N-terminal part. The central rod domain of dystrophin consists of spectrin-like repeats and serves as the second site for actin binding (Rybakova et al. 2006). Therefore, one of the functions of the DGC is to connect the extracellular matrix to the actin cytoskeleton, which is thought to provide mechanical stability to the muscle plasma membrane.
Sarcoglycans are single-pass transmembrane glycoproteins. In skeletal muscle, α-, β-, γ-, and δ-SGs are expressed, and they form a subcomplex with sarcospan in the DGC (SG-sarcospan complex) (Fig. 1). Mutations in any of the α-, β-, γ-, and δ-SG genes result in autosomal recessive limb-girdle type muscular dystrophies (Ozawa et al. 2005). A primary mutation in any one of the SG genes leads to concomitant reduction of the entire SG complex. The exact function of the SG-sarcospan complex is still unknown, but some putative functions have been proposed; it may stabilize the DGC components in the muscle membrane (Duclos et al. 1998; Straub et al. 1998); it may regulate localization of γ-filamin (Thompson et al. 2000). Using animal models and virus-gene transfer techniques, it has been demonstrated that genetic deficiencies of the SG complex can be corrected in vivo (Holt et al. 1998; Durbeej et al. 2003). Inherited cardiomyopathy can be associated with genetic defects in the DGC (Towbin 1998), and a correlation between the primary mutation of the SG gene and cardiomyopathy has been established (Melacini et al. 1999). Accordingly, the genetically engineered models of β-SG and δ-SG-null mice developed cardiomyopathy (Coral-Vasquez et al. 1999; Durbeej et al. 2000). Detailed analysis has demonstrated vascular irregularities in several tissues of β- or δ-SG-deficient animals, suggesting that the SG complex has a crucial role in the maintenance of the vascular structure to prevent the initiation of heart pathology. Importantly, the vascular dysfunction in these animals can be abolished by administration of verapamil (a calcium channel blocker with vasodilator properties), which presents a pharmacological intervention to prevent progression of cardiomyopathy (Cohn et al. 2001).
Dystroglycan and dystroglycanopathy
As mentioned, DG is a central molecule in the DGC, but so far no primary mutations of the DG gene have been found in any human disease. Although disruption of the DG gene in mice results in embryonic lethality (Williamson 1997), by using striated-muscle selective conditional DG knock-out mice, evidence for important roles of DG in the maintenance of skeletal muscle function has been provided (Cohn et al. 2002). DG is encoded by a single mRNA and post-translationally cleaved into two subunits, α- and β-DG (Fig. 2) (Ibraghimov-Beskrovnaya et al. 1992). α-DG is an extracellular subunit and functions as a receptor for several extracellular matrix proteins such as laminins, perlecan, agrin, and neurexin (Michele and Campbell 2003). α-DG is anchored on the plasma membrane through the non-covalent interaction with β-DG. β-DG is a transmembrane subunit and intracellularly interacts with dystrophin. Therefore, DG acts as a bridge molecule between the extracellular matrix and the actin–dystrophin cytoskeletons across the muscle plasma membrane (Fig. 1). α-DG undergoes extensive O-glycosylation, and biochemical studies revealed that O-glycosylation is required for the ligand-binding activity of α-DG (Ervasti and Campbell 1993). α-DG contains a Ser/Thr-rich mucin-like domain, which is a potential site for functional glycosylation (Kanagawa et al. 2004). Although the predicted molecular mass of the α-DG core peptide, following furin-dependent cleavage of the N-terminus (Kanagawa et al. 2004; Singh et al. 2004), is around 40 kDa, the size of α-DG on the SDS-gel ranges from 100–160 kDa with a tissue- or developmental-dependent manner (Barresi and Campbell 2006). Among heterogeneous groups of O-linked sugar chains on the mucin-like domain of α-DG, a unique O-mannose type sugar chain [Neu5Ac(α2-3)Gal(β1-4)GlcNAc(β1-2)Man-Ser/Thr] is reported to be involved in the ligand-binding activity (Fig. 2) (Chiba et al. 1997).

O-glycosylation of α-DG and congenital muscular dystrophies (dystroglycanopathies). α-DG is comprised of the N-terminal, the mucin-like, and the C-terminal domains. Mutations in known or putative glycosyltransferase genes have been identified in patients with congenital muscular dystrophies. In these disorders, α-DG is hypoglycosylated, and its laminin-binding activity is greatly reduced. POMGnT1 and POMT1/2 are involved in the synthesis of a unique O-mannose sugar chain. The function of fukutin and FKRP is still unknown. Molecular recognition by LARGE is essential for functional expression of α-DG. Overexpression of LARGE stimulates the synthesis of unknown O-linked sugars on the mucin-like domain
Probably the most recent significant advance in the genetic basis of muscular dystrophy is the identification of genes mutated in the O-glycosylation pathway of DG (Fig. 2). These mutations commonly result in aberrant glycosylation of DG with reduced ligand-binding activity, therefore called dystroglycanopathy (Toda et al. 2003). Abnormal glycosylation can be tested by immunofluorescence or Western blotting analysis using the monoclonal antibodies that recognize a functionally glycosylated form of α-DG (IIH6 and VIA4-1) and polyclonal antibodies recognizing the α-DG core peptide (GT20ADG). Abnormally glycosylated α-DG loses reactivity against IIH6 and VIA4-1, and shows reduced molecular weight that can be detected by Western blotting with the α-DG core antibodies (Michele et al. 2002). Dystroglycanopathy so far includes Fukuyama congenital muscular dystrophy (FCMD), muscle–eye–brain disease (MEB), Walker–Warburg syndrome (WWS), congenital muscular dystrophies type 1C (MDC1C) and 1D (MDC1D), and limb-girdle muscular dystrophy 2I (LGMD2I). In dystroglycanopathies, the reduction or loss of DG’s ligand-binding activity due to hypoglycosylation likely disrupts the DG-mediated linkage between the extracellular matrix and the dystrophin–cytoskeleton, which is believed to be a cause of this group of disease. Genes responsible for dystroglycanopathies and their determined or putative function will be presented below.
Fukuyama congenital muscular dystrophy is an autosomal recessive muscular dystrophy that is the most common congenital muscular dystrophy in Japan. In addition, FCMD is also the most common childhood muscular dystrophy next to DMD in Japan. FCMD is characterized by severe congenital muscular dystrophy, abnormal neuronal migration associated with mental retardation and epilepsy, and frequently eye abnormalities. In addition, recent study demonstrates morphologically aberrant neuromuscular junctions in FCMD skeletal muscle (Taniguchi et al. 2006). Positional cloning identified the gene responsible for FCMD, and its protein product was named fukutin (Toda et al. 1993; Kobayashi et al. 1998). The major mutation in FCMD was first identified as a 3-kb retrotransposon insertion in the 3′ noncoding region of the fukutin gene (Kobayashi et al. 1998). FCMD is the first known human disease that is caused by ancient retrotransposal integration. A high population of patients carry this ancestral insertion, which may explain the high prevalence of this disease in Japan. This genomic insertion in the FCMD gene is likely to cause a decrease in transcription of mRNA, therefore resulting in the loss of gene product function. Compound heterozygousity between retrotransposonal mutation and point-mutation results in a more severe phenotype than homozygous retrotransposonal mutation. The function of fukutin is unknown, but the protein has homologous to enzymes that modify glycolipids and glycoproteins (Aravind and Koonin 1999). In FCMD muscle, α-DG is hypoglycosylated, and the ligand binding activities are dramatically decreased (Michele et al. 2002), which indicates that fukutin is involved in the glycosylation pathway of DG. Generation of fukutin chimeric mice demonstrated the essential role of fukutin-dependent glycosylation of DG in the maintenance of muscle integrity, cortical histiogenesis, and ocular development (Takeda et al. 2003). On the other hand, targeted homozygous mutation of the fukutin gene in mice leads to embryonic lethality prior to development of skeletal muscle and mature neurons, and detailed analyses on the mutant embryos suggest that fukutin is necessary for the maintenance of the basement membrane function, which is required for early embryonic development (Kurahashi et al. 2005).
A homologue for fukutin was identified based on its sequence homology, and the gene product was termed fukutin-related protein (FKRP) (Brockington et al. 2001a). According to its amino acid sequence and cellular localization study (Esapa et al. 2002), FKRP is thought to be a glycosyltransferase, but the enzyme activity has not been determined. Mutations in the FKRP can be detected in a broad patient population. The two distinct phenotypes can be categorized into congenital muscular dystrophy 1C (MDC1C) and milder limb-girdle muscular dystrophy 2I (LGMD2I) (Brockington et al. 2001b). MDC1C displays a profound depletion of α-DG functional glycosylation, whereas LGMD2I shows a moderate reduction of α-DG glycosylation (Brown et al. 2004). Moreover, individuals with milder LGMD2I show a variable, but subtle alteration in α-DG glycosylation (Brown et al. 2004). These may indicate a correlation between the residual α-DG glycosylation level and the disease severity.
Muscle–eye–brain disease is a severe autosomal recessive disease characterized by congenital muscular dystrophy, ocular abnormalities, and brain malformation and is seen mainly in Finland. As with FCMD, hypoglycosylation of α-DG is commonly seen in MEB patient cells (Kano et al. 2002; Michele et al. 2002; Barresi et al. 2004). The gene responsible for MEB is the POMGnT1, which codes a UDP-GlcNAc:Manα-O β1,2-N-acetylglucosaminyltransferase. The GlcNAcβ1-2Man linkage in the O-mannose sugar chain on α-DG is unique to mammals and had been assumed to be catalyzed by POMGnT. In 2001, Yoshida et al. reported finding the POMGnT1 gene by EST cloning based on a sequence homologous to human GnT-1, which synthesizes GlcNAcβ1-2Man linkage in N-glycans (Yoshida et al. 2001). Subsequently, the cloned protein was confirmed to have POMGnT activity. Screening of the whole coding region and the exon/intron flanking sequence of the POMGnT1 gene identified mutations in MEB that included a combination of missense, non-sense, and frameshift mutations, and the mutated POMGnT1 proteins showed reduced enzyme activities (Yoshida et al. 2001; Manya et al. 2003). This genetic and biochemical information contributed to the development of an enzymatic assay for MEB diagnostic testing that uses commercially available reagents (Zhang et al. 2003).
Walker–Warburg syndrome is the most severe dystroglycanopathy, with most patients living only a year or less. Similar to FCMD and MEB, this disease is characterized by congenital muscular dystrophy, structural brain defects, and eye malformations. To unravel the genetic basis of WWS, a genome-wide linkage analysis and a candidate-gene approach were performed, which identified mutations in the gene encoding O-mannosyltransferase 1 (POMT1) in 6 of 30 unrelated WWS cases (Beltran-Valero de Bernabe et al. 2002). Recently, mutations of the POMT2, a homologue of POMT1 with 33% of identity, in WWS patients were also reported (van Reeuwijk et al. 2005). POMT1 is homologous to members of the family of protein O-mannosyltransferases (PMT) in yeast. In yeast, PMTs catalyze the transfer of a mannosyl residue from dolichyl phosphate mannose to Ser/Thr residues of proteins (Strahl-Bolsinger et al. 1999), therefore it had been hypothesized that POMT1 might catalyze the first step in O-mannosyl glycan synthesis of α-DG. The group of Tamao Endo developed a method to detect the enzymatic activity of POMT in mammalian cells and revealed that an active POMT1/POMT2 complex is required for expression of O-mannosyltransferase activity, which adds a mannose residue directly to the polypeptide backbone of α-DG (Manya et al. 2004; Akasaka-Manya et al. 2006). WWS shows a high degree of genetic heterogeneity. Fukutin and FKRP have also been implicated in WWS; however, these genetic data still only account for a minority of cases (Muntoni and Voit 2004). Other unidentified genes may underlie WWS and glycosylation of DG.
The Largemyd mouse is a spontaneous animal model for dystroglycanopathy. As in human dystroglycanopathy, α-DG in Largemyd mice is hypoglycosylated with reduced ligand-binding activities (Grewal et al. 2001; Michele et al. 2002). A mutation in the Largemyd mouse was identified by positional cloning in the gene LARGE (Grewal et al. 2001). The mutation is an intragenic deletion, which causes a frame shift resulting in premature termination. The Large gene encodes a protein with a transmembrane domain followed by a coiled-coil domain and two DxD-containing putative catalytic domains (Peyrard et al. 1999). The gene product, LARGE, is hypothesized to be a glycosyltransferase, but the enzyme activities have not been determined yet. Adenoviral LARGE gene transfer in Largemyd restores DG functional expression and prevents the progression of muscular dystrophy (Barresi et al. 2004). Overexpression of LARGE has been shown to cause hyperglycosylation of DG in culture cells and mice (Kanagawa et al. 2004; Barresi et al. 2004), confirming that LARGE is involved in the regulation of the DG functional glycosylation pathway. A muscular dystrophy patient with mental retardation who has a missense mutation and a 1-bp insertion in the LARGE has been reported, and this disorder has been named MDC1D (Longman et al. 2003). Examination of the brains of Largemyd mice revealed abnormal glycosylation of α-DG, leading to the functional loss of ligand-binding activity, abnormal cerebral cortical layering resembling cobblestone lissencephaly seen in human dystroglycanopathies, and defects in cerebellar granule cell migration (Michele et al. 2002; Kanagawa et al. 2005). Selective deletion of DG in the mouse brain also demonstrated striking abnormalities resembling those found in the Largemyd mice and dystroglycanopathy patients (Moore et al. 2002). Using these model animals, brain phenotypes in dystroglycanopathies, such as disruption of the basement membrane, abnormal neuronal migration, and brain malformation, can be explained by a loss of function of α-DG.
It has been shown that LARGE binds to the N-terminal domain of α-DG during the maturational processing of DG (Kanagawa et al. 2004), probably in the Golgi apparatus considering the cellular location of LARGE (Grewal et al. 2005; Brockington et al. 2005). This molecular recognition by LARGE is proposed as an essential event in order to express functional DG, because without the LARGE-recognition motif, DG is not functionally expressed in vivo (Kanagawa et al. 2004). The recently identified LARGE-relative protein, LARGE2 (also known as GYLTL1B), can stimulate DG hyperglycosylation and recognizes the N-terminal domain of α-DG (Fujimura et al. 2005; Grewal et al. 2005; Brockington et al. 2005). As mentioned, there is much evidence to support that LARGE is involved in the synthesis of the sugar-chain on DG (Barresi et al. 2004; Fujimura et al. 2005; Patnaik and Stanley 2005). Interestingly, overexpression of LARGE can produce hyperglycosylated DG with increased ligand-binding activity in cells from genetically distinct diseases with defective glycosylation of α-DG (FCMD, MEB, and WWS) (Barresi et al. 2004). Although the detailed mechanism of LARGE-dependent hyperglycosylation of α-DG is still unclear, the fact that overexpression of LARGE can produce a DG species with higher ligand-binding activity may lead to a novel strategy to treat dystroglycanopathies by bypassing the alternative DG-laminin linkage. Thus, glycotherapies and treatments aimed at modulating the expression or the activity of LARGE may be a future therapeutic option for the whole group of glycosyltransferase-deficient muscular dystrophies.
Extracellular matrix proteins and receptors
Skeletal muscle fibers are surrounded by an extracellular matrix called the basement membrane. The basement membrane is composed of two layers. The basal lamina is a felt-like layer that is directly linked to the plasma membrane. Another layer is the external and fibrillar reticular lamina (Sanes 2003). The basement membrane is believed to be involved in lateral force transmission of contractile force generated by the myofibrils and provides the tensile strength of the muscle structure. Accordingly, genetic evidence has revealed that congenital muscular dystrophies can arise from the loss of components in the extracellular matrix proteins and their membrane receptors.
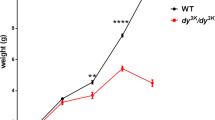
Laminin-2 (merosin, lamininα2β1γ1) is a major component of the skeletal muscle basal lamina (Fig. 1). Laminin-2 presents the forms of a cross-shaped heterotrimer through the association of an α2-, a β1-, and a γ1-chain, each of which is encoded by separate genes. Laminin-2 forms polymers through the short arm regions, and this laminin network adheres to the plasma membrane through the laminin globular domain in the α2-chain (Colognato and Yurchenco 2000). DG and integrin are well-characterized receptors of laminin-2 (Michele and Campbell 2003; Mayer 2003). A major class of congenital muscular dystrophy (MDC1A) is caused by mutations in the LAMA2 gene that encodes the laminin α2-chain (Helbling-Leclerc et al. 1995). Several mouse strains, spontaneous dy (Xu et al. 1994a) and dy2J (Xu et al. 1994b) mice and the genetically engineered dyW (Miyagoe et al. 1997) and dy3K (Kuang et al. 1998) mice, serve as animal models of the disease. Ultrastructural studies have shown an abnormal basement membrane structure around the fibers of MDC1A patients (Minetti et al. 1996) and the dy mouse (Xu et al. 1994a). The dy2J mouse presents an in-frame deletion in the short arm region of the α2-chain, demonstrating that an incomplete laminin-polymer formation can lead to muscular dystrophy (Colognato and Yurchenco 1999). Thus, the laminin polymer scaffold appears to be necessary for the organization of the basement membrane and the interaction between the matrix and cell membranes, both of which are essential for maintenance of the skeletal muscle functions. Interestingly, mini-agrin, which retains binding affinities to both DG and laminin α4- and α5-chains (up-regulated α-chains in laminin α2-deficient muscle), is shown to be able to compensate for loss of the laminin α2-chain in dyW mice by establishing an alternative DG-matrix linkage (Moll et al. 2001). Laminin–receptor interaction is also thought to be involved in the apoptotic pathway that may underlie the pathogenesis of muscular dystrophy because of the increased signs of apoptosis that were reported in laminin-2-deficient mice and human patients (Miyagoe et al. 1997; Hayashi et al. 2001). Genetic interventions designed to inhibit the mitochondrial apoptotic pathway demonstrate an improvement of the disease pathology and lifespan of laminin-2-deficient mice (Girgenrath et al. 2004; Dominov et al. 2005), which suggests the possibility of using anti-apoptosis therapy for muscular dystrophies.
Integrin α7β1 provides a linkage between laminin-2 and the cytoskeleton as well as the DGC (Fig. 1). Mice carrying an inactivated integrin α7 gene (ITGA7) were shown to develop a mild, but progressive muscular dystrophy (Mayer et al. 1997). Only Japanese patients so far have been identified with a primary integrin α7 deficiency and clinically classified as having congenital myopathy with a delayed motor milestone (Hayashi et al. 1998). The integrin α7 deficiency was reported to be caused by splice mutations and deletions, leading to frameshifts, and/or marked reduction of ITGA7 mRNA. In the integrin α7-deficient mice, the myotendinous junction, the primary site of force transmission between the muscle and the tendon, was severely disrupted, indicating an important role of integrin α7 in the organization and/or maintenance of the myotendinous junction (Mayer et al. 1997; Nawrotzki et al. 2003). The generation of integrin α7/dystrophin- or integrin α7/sarcoglycan-double mutant mice demonstrates an essential role for integrin α7 in the maintenance of the DGC-deficient muscle plasma membrane (Allikian et al. 2004; Guo et al. 2006; Rooney et al. 2006). Overexpression of integrin α7 in dystrophin/utrophin double mutant mice, a mouse model for DMD, ameliorates the muscle disease phenotype (Burkin et al. 2001). These studies suggest a possibility that integrin α7 may be a target molecule to compensate for defects of the DGC by reinforcing the laminin matrix–cytoskeleton linkage.
Collagen VI is an extracellular matrix protein composed of three chains, α1, α2, and α3, encoded by the COL6A1, the COL6A2, and the COL6A3, respectively, and the trimer collagen VI eventually assembles into microfilament network (Zhang et al. 2002). Collagen VI localizes in the reticular layer of the basement membrane, perimysium and endomysium (Fig. 1). The collagen VI network interacts with the fibronectin networks, biglycan, and collagen IV (Jimenez-Mallebrera et al. 2005). Mutations in the collagen VI genes are known to cause Ullrich syndrome and Bethlem myopathy (Camacho-Vanegas et al. 2001; Demir et al. 2002; Pan et al. 2003). Absence of collagen VI microfibrils from the area adjacent to the muscle basal lamina in Ullrich syndrome patients has been reported (Ishikawa et al. 2002; Niiyama et al. 2002). The data support a hypothesized role of collagen VI in anchoring the muscle basement membrane to the ECM. In addition, an in vitro study using fibroblast culture indicates that collagen VI may be important for the organization of the components of the ECM (Sabatelli et al. 2001). The COL6A knock-out mice show reduced contractile force and disturbed intracellular calcium homeostasis (Bonaldo et al. 1998). These studies indicate a role of collagen VI in anchoring the basal lamina to the reticular layer for maintenance of muscle function. The loss of contractile strength of the COL6A mutant mice appeared to be associated with ultrastructural alterations of the sarcoplasmic reticulum and mitochondria and spontaneous apoptosis. Interestingly, these defects were rescued in vitro by adding collagen VI and in vivo by cyclosporine treatment, which may lead to pharmacological intervention in the diseases (Irwin et al. 2003).
Therapeutic approaches
A simple way to cure muscular dystrophies is by replacing the defective gene in the disease with the normal version of the gene. Many studies aimed at developing a therapeutic strategy for DMD, which is the most frequent muscular dystrophy, have been undertaken by using the model mdx mice. However, the size of the dystrophin gene has led to difficulties in vector construction and efficiency of gene delivery. This issue was circumvented by the use of truncated “mini” dystrophins with functional capacities near that of full-length dystrophin. Viral or nonviral delivery of the mini dystrophins has been shown to rescue the dystrophic muscle in mdx mice (for a review, see van Deutekom and van Ommen 2003). The mdx mouse and a minority of DMD patients have a premature stop codon in the dystrophin gene. Interestingly, the aminoglycoside gentamicin acts as a suppressor of this stop codon mutation, allowing read-through of the dystrophin cDNA. It has been reported that mdx mice treated with gentamicin have an increased dystrophin expression level and show improvement in the disease phenotype (Barton-Davis et al. 1999). In addition, anti-sense oligonucleotides have emerged as a potential to induce exon skipping to restore a disrupted reading frame or omit a nonsense mutation (Lu et al. 2003).
Functional bypassing of the missing protein by activating an alternative pathway is a relatively new strategy for muscular dystrophy treatment. As already mentioned, reinforcement of the linkage between DG and matrix proteins by LARGE (Barresi et al. 2004) or mini agrin expression (Moll et al. 2001) is an intriguing strategy for dystroglycanopathies and laminin-deficient muscular dystrophies, respectively. As for DMD, utrophin is a target protein for the compensation of dystrophin defects. Utrophin is a homologue of dystrophin (Tinsley et al. 1992) and is upregulated in the regenerating muscle of DMD patients, which has led to the hypothesis that utrophin might have a complementary role in dystrophin-deficient muscle (Mizuno et al. 1993). Accordingly, transgenic or viral expression of full-length or mini utrophin in mdx mice improves the disease severity (van Deutekom and van Ommen 2003). Recently, the administration of L-arginine or transgenic expression of ADAM12 (a disintegrin and metalloprotease) was reported to enhance utrophin expression (Chaubourt et al. 2002; Moghadaszadeh 2003). Understanding the molecular basis of utrophin upregulation may lead to the design of new strategies to treat DMD.
Although these strategies suggest potentially exciting ways to treat muscular dystrophies, there are a number of hurdles to overcome for their eventual use in humans. Gene therapy will require the development of methods to deliver the viral vector effectively to all muscles of the body and also has the problem of stimulating an immune response. Pharmaceutical treatments may slow the progression of the disease, but they do not substantially affect the long-term outcome. Stem cell therapy may potentially overcome these issues and be a major focus in the future for the management of a broad range of muscular dystrophies. Recent studies demonstrated that bone marrow stromal cells (Dezawa et al. 2005) or muscle-derived stem cells (Tamaki et al. 2005) are able to generate muscle cells and can repair muscle degeneration in vivo. Reconstitution of the healthy gene into such stem cells prepared from the patients and their retransplantation might be a new potential strategy to treat muscular dystrophies.
Conclusion
With the combination of biochemistry, muscle cell biology, and the use of modeling animals, molecular genetic-based information has shed light on understanding the pathomechanism of muscular dystrophies. As reviewed, primary genetic or the secondary functional disruption of the matrix–plasma membrane linkage appears to be the cause of several forms of muscular dystrophies. The mechanical property of skeletal muscle is maintained by contractile elements and elastic elements, which are provided by the sarcomere and the extracellular matrix, respectively. In addition, lateral transmission of the contractile force is mediated by matrix–receptor interaction. Thus, based on the fact that the matrix–cytoskeleton linkage is a key for maintenance of skeletal muscle function, several therapeutic strategies have been proposed as introduced in this review and by others. Hopefully, such approaches will contribute to a greater understanding of the disease etiology and lead to appropriate therapeutic strategies to treat muscular dystrophies.
References
Akasaka-Manya K, Manya H, Nakajima A, Kawakita M, Endo T (2006) Physical and functional association of human protein o-mannosyltransferases 1 and 2. J Biol Chem 281:19339–19345
Allikian MJ, Hack AA, Mewborn S, Mayer U, McNally EM (2004) Genetic compensation for sarcoglycan loss by integrin alpha7beta1 in muscle. J Cell Sci 117:3821–3830
Aravind L, Koonin EV (1999) The fukutin protein family-predicted enzymes modifying cell-surface molecules. Curr Biol 9:R836–837
Barresi R, Campbell KP (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 119:199–207
Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS, Moore SA, Zhang W, Schachter H, Dumanski JP, Cohn RD, Nishino I, Campbell KP (2004) LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med 10:696–703
Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL (1999) Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104:375–381
Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG (2002) Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am J Hum Genet 71:1033–1043
Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM (1998) Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 7:2135–2140
Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, Muntoni F (2001a) Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 69:1198–1209
Brockington M, Torelli S, Prandini P, Boito C, Dolatshad NF, Longman C, Brown SC, Muntoni F (2005) Localization and functional analysis of the LARGE family of glycosyltransferases: significance for muscular dystrophy. Hum Mol Genet 14:657–665
Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, Herrmann R, Anderson LV, Bashir R, Burgunder JM, Fallet S, Romero N, Fardeau M, Straub V, Storey G, Pollitt C, Richard I, Sewry CA, Bushby K, Voit T, Blake DJ, Muntoni F (2001b) Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 10:2851–2859
Brown SC, Torelli S, Brockington M, Yuva Y, Jimenez C, Feng L, Anderson L, Ugo I, Kroger S, Bushby K, Voit T, Sewry C, Muntoni F (2004) Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 164:727–737
Burkin DJ, Wallace GQ, Nicol KJ, Kaufman DJ, Kaufman SJ (2001) Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol 152:1207–1218
Camacho-Vanegas O, Bertini E, Zhang RZ, Petrini S, Minosse C, Sabatelli P, Giusti B, Chu ML, Pepe G (2001) Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA 98:7516–7521
Campbell KP, Kahl SD (1989) Association of dystrophin and an integral membrane glycoprotein. Nature 338:259–262
Chaubourt E, Voisin V, Fossier P, Baux G, Israel M, De La Porte S (2002) Muscular nitric oxide synthase (muNOS) and utrophin. J Physiol Paris 96:43–52
Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T (1997) Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha–dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha–dystroglycan with laminin. J Biol Chem 272:2156–2162
Cohn RD, Durbeej M, Moore SA, Coral-Vazquez R, Prouty S, Campbell KP (2001) Prevention of cardiomyopathy in mouse models lacking the smooth muscle sarcoglycan-sarcospan complex. J Clin Invest 107:R1–R7
Cohn RD, Henry MD, Michele DE, Barresi R, Saito F, Moore SA, Flanagan JD, Skwarchuk MW, Robbins ME, Mendell JR, Williamson RA, Campbell KP (2002) Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell 110:639–648
Colognato H, Yurchenco PD (1999) The laminin alpha2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers. Curr Biol 9:1327–1330
Colognato H, Yurchenco PD (2000) Form and function: the laminin family of heterotrimers. Dev Dyn 218:213–234
Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP (1999) Disruption of the sarcoglycan–sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 98:465–474
Dalkilic I, Kunkel LM (2003) Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 13:231–238
Demir E, Sabatelli P, Allamand V, Ferreiro A, Moghadaszadeh B, Makrelouf M, Topaloglu H, Echenne B, Merlini L, Guicheney P (2002) Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet 70:1446–1458
Dezawa M, Ishikawa H, Itokazu Y, Yoshihara T, Hoshino M, Takeda S, Ide C, Nabeshima Y (2005) Bone marrow stromal cells generate muscle cells and repair muscle degeneration. Science 309:314–317
Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB (2005) Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet 14:1029–1040
Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA, Campbell KP (2000) Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell 5:141–151
Durbeej M, Sawatzki SM, Barresi R, Schmainda KM, Allamand V, Michele DE, Campbell KP (2003) Gene transfer establishes primacy of striated vs. smooth muscle sarcoglycan complex in limb-girdle muscular dystrophy. Proc Natl Acad Sci USA 100:8910–8915
Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, Holt KH, Lim LE, Sanes JR, Davidson BL, Faulkner JA, Williamson R, Campbell KP (1998) Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol 142:1461–1471
Ervasti JM, Campbell KP (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122:809–823
Esapa CT, Benson MA, Schroder JE, Martin-Rendon E, Brockington M, Brown SC, Muntoni F, Kroger S, Blake DJ (2002) Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet 11:3319–3331
Fujimura K, Sawaki H, Sakai T, Hiruma T, Nakanishi N, Sato T, Ohkura T, Narimatsu H (2005) LARGE2 facilitates the maturation of alpha–dystroglycan more effectively than LARGE. Biochem Biophys Res Commun 329:1162–1171
Girgenrath M, Dominov JA, Kostek CA, Miller JB (2004) Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest 114:1635–1639
Grewal PK, Holzfeind PJ, Bittner RE, Hewitt JE (2001) Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat Genet 28:151–154
Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE (2005) Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15:912–923
Guo C, Willem M, Werner A, Raivich G, Emerson M, Neyses L, Mayer U (2006) Absence of alpha7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum Mol Genet 15:989–998
Hayashi YK, Chou FL, Engvall E, Ogawa M, Matsuda C, Hirabayashi S, Yokochi K, Ziober BL, Kramer RH, Kaufman SJ, Ozawa E, Goto Y, Nonaka I, Tsukahara T, Wang JZ, Hoffman EP, Arahata K (1998) Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 19:94–97
Hayashi YK, Tezak Z, Momoi T, Nonaka I, Garcia CA, Hoffman EP, Arahata K (2001) Massive muscle cell degeneration in the early stage of merosin-deficient congenital muscular dystrophy. Neuromuscul Disord 11:350–359
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé F, Schwartz K, Fardeau M, Tryggvason K, Guicheney P (1995) Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet 11:216–218
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Holt KH, Lim LE, Straub V, Venzke DP, Duclos F, Anderson RD, Davidson BL, Campbell KP (1998) Functional rescue of the sarcoglycan complex in the BIO 14.6 hamster using delta-sarcoglycan gene transfer. Mol Cell 1:841–848
Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP (1992) Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355:696–702
Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P, Bonaldo P (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35:367–371
Ishikawa H, Sugie K, Murayama K, Ito M, Minami N, Nishino I, Nonaka I (2002) Ullrich disease: collagen VI deficiency: EM suggests a new basis for muscular weakness. Neurology 59:920–923
Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F (2005) Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 62:809–823
Kanagawa M, Michele DE, Satz JS, Barresi R, Kusano H, Sasaki T, Timpl R, Henry MD, Campbell KP (2005) Disruption of perlecan binding and matrix assembly by post-translational or genetic disruption of dystroglycan function. FEBS Lett 579:4792–4796
Kanagawa M, Saito F, Kunz S, Yoshida-Moriguchi T, Barresi R, Kobayashi YM, Muschler J, Dumanski JP, Michele DE, Oldstone MB, Campbell KP (2004) Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell 117:953–964
Kano H, Kobayashi K, Herrmann R, Tachikawa M, Manya H, Nishino I, Nonaka I, Straub V, Talim B, Voit T, Topaloglu H, Endo T, Yoshikawa H, Toda T (2002) Deficiency of alpha–dystroglycan in muscle–eye–brain disease. Biochem Biophys Res Commun 291:1283–1286
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394:388–392
Kuang W, Xu H, Vachon PH, Liu L, Loechel F, Wewer UM, Engvall E (1998) Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest 102:844–852
Kurahashi H, Taniguchi M, Meno C, Taniguchi Y, Takeda S, Horie M, Otani H, Toda T (2005) Basement membrane fragility underlies embryonic lethality in fukutin-null mice. Neurobiol Dis 19:208–217
Lapidos KA, Kakkar R, McNally EM (2004) The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94:1023–1031
Longman C, Brockington M, Torelli S, Jimenez-Mallebrera C, Kennedy C, Khalil N, Feng L, Saran RK, Voit T, Merlini L, Sewry CA, Brown SC, Muntoni F (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 12:2853–2861
Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, Xue SA, Fletcher S, Partridge TA, Wilton SD (2003) Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 9:1009–1014
Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, Margolis RU, Endo T (2004) Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA 101:500–505
Manya H, Sakai K, Kobayashi K, Taniguchi K, Kawakita M, Toda T, Endo T (2003) Loss-of-function of an N-acetylglucosaminyltransferase, POMGnT1, in muscle–eye–brain disease. Biochem Biophys Res Commun 306:93–97
Mayer U (2003) Integrins: redundant or important players in skeletal muscle? J Biol Chem 278:14587–14590
Mayer U, Saher G, Fassler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Poschl E, von der Mark K (1997) Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet 17:318–323
Melacini P, Fanin M, Duggan DJ, Freda MP, Berardinelli A, Danieli GA, Barchitta A, Hoffman EP, Dalla Volta S, Angelini C (1999) Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve 22:473–479
Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP (2002) Post-translational disruption of dystroglycan–ligand interactions in congenital muscular dystrophies. Nature 418:417–422
Michele DE, Campbell KP (2003) Dystrophin–glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem 278:15457–15460
Minetti C, Bado M, Morreale G, Pedemonte M, Cordone G (1996) Disruption of muscle basal lamina in congenital muscular dystrophy with merosin deficiency. Neurology 46:1354–1358
Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S (1997) Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene a new model of merosin (laminin 2)-deficient congenital muscular dystrophy FEBS Lett 415:33–39
Mizuno Y, Nonaka I, Hirai S, Ozawa E (1993) Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD-carriers and control subjects. J Neurol Sci 119:43–52
Moghadaszadeh B, Albrechtsen R, Guo LT, Zaik M, Kawaguchi N, Borup RH, Kronqvist P, Schroder HD, Davies KE, Voit T, Nielsen FC, Engvall E, Wewer UM (2003) Compensation for dystrophin-deficiency: ADAM12 overexpression in skeletal muscle results in increased alpha 7 integrin, utrophin and associated glycoproteins. Hum Mol Genet 12:2467–2479
Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, Muller U, Ruegg MA (2001) An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 413:302–307
Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM (1986) Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323:646–650
Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418:422–425
Morris GE (2000) Nuclear proteins and cell death in inherited neuromuscular disease. Neuromuscul Disord 10: 217–227
Muntoni F, Voit T (2004) The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 14:635–649
Nawrotzki R, Willem M, Miosge N, Brinkmeier H, Mayer U (2003) Defective integrin switch and matrix composition at alpha 7-deficient myotendinous junctions precede the onset of muscular dystrophy in mice. Hum Mol Genet 12:483–95
Niiyama T, Higuchi I, Suehara M, Hashiguchi T, Shiraishi T, Nakagawa M, Arimura K, Maruyama I, Osame M (2002) Electron microscopic abnormalities of skeletal muscle in patients with collagen VI deficiency in Ullrich’s disease. Acta Neuropathol (Berl) 104:67–71
Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M (2005) Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 32:563–576
Patnaik SK, Stanley P (2005) Mouse large can modify complex N- and mucin O-glycans on alpha–dystroglycan to induce laminin binding. J Biol Chem 280:20851–20859
Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML (2003) New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet 73:355–369
Peyrard M, Seroussi E, Sandberg-Nordqvist AC, Xie YG, Han FY, Fransson I, Collins J, Dunham I, Kost-Alimova M, Imreh S, Dumanski JP (1999) The human LARGE gene from 22q12.3–q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc Natl Acad Sci USA 96:598–603
Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ, Burkin DJ (2006) Severe muscular dystrophy in mice that lack dystrophin and {alpha}7 integrin. J Cell Sci 119:2185–2195
Rybakova IN, Humston JL, Sonnemann KJ, Ervasti JM (2006) Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem 281:9996–10001
Sabatelli P, Bonaldo P, Lattanzi G, Braghetta P, Bergamin N, Capanni C, Mattioli E, Columbaro M, Ognibene A, Pepe G, Bertini E, Merlini L, Maraldi NM, Squarzoni S (2001) Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts. Matrix Biol 20:475–486
Sanes JR (2003) The basement membrane/basal lamina of skeletal muscle. J Biol Chem 278:12601–12604
Singh J, Itahana Y, Knight-Krajewski S, Kanagawa M, Campbell KP, Bissell MJ, Muschler J (2004) Proteolytic enzymes and altered glycosylation modulate dystroglycan function in carcinoma cells. Cancer Res 64:6152–6159
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN (2003) Human Gene Mutation Database (HGMD®): 2003 update. Hum Mutat 21:577–581
Strahl-Bolsinger S, Gentzsch M, Tanner W (1999) Protein O-mannosylation. Biochim Biophys Acta 1426:297–307
Straub V, Duclos F, Venzke DP, Lee JC, Cutshall S, Leveille CJ, Campbell KP (1998) Molecular pathogenesis of muscle degeneration in the delta–sarcoglycan-deficient hamster. Am J Pathol 153:1623–1630
Takeda S, Kondo M, Sasaki J, Kurahashi H, Kano H, Arai K, Misaki K, Fukui T, Kobayashi K, Tachikawa M, Imamura M, Nakamura Y, Shimizu T, Murakami T, Sunada Y, Fujikado T, Matsumura K, Terashima T, Toda T (2003) Fukutin is required for maintenance of muscle integrity, cortical histiogenesis and normal eye development. Hum Mol Genet 12:1449–1459
Tamaki T, Uchiyama Y, Okada Y, Ishikawa T, Sato M, Akatsuka A, Asahara T (2005) Functional recovery of damaged skeletal muscle through synchronized vasculogenesis, myogenesis, and neurogenesis by muscle-derived stem cells. Circulation 112:2857–2866
Taniguchi M, Kurahashi H, Noguchi S, Fukudome T, Okinaga T, Tsukahara T, Tajima Y, Ozono K, Nishino I, Nonaka I, Toda T (2006) Aberrant neuromuscular junctions and delayed terminal muscle fiber maturation in alpha–dystroglycanopathies. Hum Mol Genet 15:1279–1289
Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S, Kunkel LM (2000) Filamin 2 (FLN2): a muscle-specific sarcoglycan interacting protein. J Cell Biol 148:115–126
Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE (1992) Primary structure of dystrophin-related protein. Nature 360:591–593
Toda T, Kobayashi K, Takeda S, Sasaki J, Kurahashi H, Kano H, Tachikawa M, Wang F, Nagai Y, Taniguchi K, Taniguchi M, Sunada Y, Terashima T, Endo T, Matsumura K (2003) Fukuyama-type congenital muscular dystrophy (FCMD) and alpha–dystroglycanopathy. Congenit Anom (Kyoto) 43:97–104
Toda T, Segawa M, Nomura Y, Nonaka I, Masuda K, Ishihara T, Suzuki M, Tomita I, Origuchi Y, Ohno K, Misugi N, Sasaki Y, Takada K, Kawai M, Otani K, Murakami T, Saito K, Fukuyama Y, Shimizu T, Kanazawa I, Nakamura Y (1993) Localization of a gene for Fukuyama type congenital muscular dystrophy to chromosome 9q31–33. Nat Genet 5:283–286
Towbin JA (1998) The role of cytoskeletal proteins in cardiomyopathies. Curr Opin Cell Biol 10:131–139
van Deutekom JC, van Ommen GJ (2003) Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet 4:774–783
van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, Huynen MA, Verrips A, Walsh CA, Barth PG, Brunner HG, van Bokhoven H (2005) POMT2 mutations cause alpha–dystroglycan hypoglycosylation and Walker–Warburg syndrome. J Med Genet 42:907–912
Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP (1997) Dystroglycan is essential for early embryonic development: disruption of Reichert’s membrane in Dag1-null mice. Hum Mol Genet 6:831–841
Xu H, Christmas P, Wu XR, Wewer UM, Engvall E (1994a) Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA 91:5572–5576
Xu H, Wu XR, Wewer UM, Engvall E (1994b) Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet 8:297–302
Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1:717–724
Yoshida M, Ozawa E (1990) Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem (Tokyo) 108:748–752
Zhang RZ, Sabatelli P, Pan TC, Squarzoni S, Mattioli E, Bertini E, Pepe G, Chu ML (2002) Effects on collagen VI mRNA stability and microfibrillar assembly of three COL6A2 mutations in two families with Ullrich congenital muscular dystrophy. J Biol Chem 277:43557–43564
Zhang W, Vajsar J, Cao P, Breningstall G, Diesen C, Dobyns W, Herrmann R, Lehesjoki AE, Steinbrecher A, Talim B, Toda T, Topaloglu H, Voit T, Schachter H (2003) Enzymatic diagnostic test for muscle–eye–brain type congenital muscular dystrophy using commercially available reagents. Clin Biochem 36:339–344
Acknowledgement
We would like to thank Dr. Kevin Campbell, Dr. Aaron Beedle, Dr. Yvonne Kobayashi, and Dr. Helena Popiel for critical reading of the manuscript and helpful suggestions. This work was supported by the 21st Century COE program from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kanagawa, M., Toda, T. The genetic and molecular basis of muscular dystrophy: roles of cell–matrix linkage in the pathogenesis. J Hum Genet 51, 915–926 (2006). https://doi.org/10.1007/s10038-006-0056-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0056-7
Keywords
This article is cited by
-
Comparison of telerehabilitation versus home-based video exercise in patients with Duchenne muscular dystrophy: a single-blind randomized study
Acta Neurologica Belgica (2022)
-
The linkage between inflammation and fibrosis in muscular dystrophies: The axis autotaxin–lysophosphatidic acid as a new therapeutic target?
Journal of Cell Communication and Signaling (2021)
-
Efficient engraftment of pluripotent stem cell-derived myogenic progenitors in a novel immunodeficient mouse model of limb girdle muscular dystrophy 2I
Skeletal Muscle (2020)
-
Skeletal, cardiac, and respiratory muscle function and histopathology in the P448Lneo− mouse model of FKRP-deficient muscular dystrophy
Skeletal Muscle (2018)
-
Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice
Nature Communications (2018)
