
Abstract
The solid oxide fuel cell symmetrical cells with porous (La1-x Sr x ) y CoO3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ electrodes for intermediate temperature applications have been studied under electrochemical polarization and synthetic air + H2O vapor (so called moisturized cathode gas) feeding conditions using high-temperature electrochemical in situ X-ray diffraction method, chronoamperometry, cyclic voltammetry, and impedance spectroscopy methods. Changes in the lattice parameters and electrochemical activity of La0.6Sr0.4CoO3−δ , (La0.6Sr0.4)1.01CoO3−δ , (La0.6Sr0.4)0.99CoO3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ were calculated depending on the temperature (T), electrode potential (E), and oxygen partial pressure (p O2) applied. Influence of H2O vapor in synthetic air on (La1-x Sr x ) y CoO3−δ parameters was irreversible and continued expansion of (La1-x Sr x ) y CoO3−δ lattice observed in H2O vapor + synthetic air feeding conditions has been observed continuously after changing wet cathode gas to dry oxygen. Nearly, reversible behavior of La0.6Sr0.4Co0.2Fe0.8O3−δ lattice has been established and for La0.6Sr0.4Co0.2Fe0.8O3−δ, the cell volume and polarization resistance started to decrease after changing humidified synthetic air to dry synthetic air in the cathode compartment. For the slightly cationic deficient (La0.6Sr0.4)0.99CoO3−δ , the cathode structure is more stable and the electroreduction of the oxygen was faster. Detailed comparison of experimental data demonstrates that the dependence of the crystallographic parameters on the electrode potential and temperature applied is in a good agreement with the electrochemical impedance spectroscopy data, indicating that the electrocatalytic activity of the cathode decreases with the rise of Fe ion concentration in B-position of LSCF cathode.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Solid oxide fuel cell (SOFC) technology plays more and more important role in high-efficiency energetic applications being attractive for early energy market [1–7]. The high energy and power density of SOFC and good electrical efficiency dignify SOFC technology as a potential replacement for traditional combustion engine based solutions and even for traditional power plants. La0.6Sr0.4CoO3−δ (LSC) cathode as an excellent mixed electron and ion conductor in SOFC family, within intermediate temperature region, has a high resistivity against high temperature degradation and reasonable chemical suitability with the ceria based electrolytes [1–6]. Increasing interest in the operation of SOFC at temperatures considerably lower than 1000 °C, has raised the need for theoretical studies for a better understanding of the factors determining the time stability and limiting parameters determining the electrical performance of the electrodes at T ≤ 680 °C.
Strontium substituted lanthanum cobaltite La1-x Sr x CoO3−δ is a perovskite-type oxide with high electronic and oxide ion conductivity at moderate and high temperatures and is therefore considered for use in high temperature devices such as ceramic membranes, electrolysers, and fuel cells. The mixed conducting nature of LSC is believed to account for the lower oxygen electroreduction overpotential, when used as a SOFC-cathode, explained by expansion of the catalytically active area of the electrode beyond the triple phase boundary region [8].
Based on the literature data [1–6], the catalytic activity and structural stability of cathode are determined by the chemical composition of the cathode material. LSCF cathode is less active against oxygen electroreduction process, but the Fe cation stabilizes the crystallographic structure of electrode, increasing the resistance against chemical reagents (including H2O), thus, increasing the life time and durability of SOFC cells based on LSCF. However, it should be noted, that the polarization resistance of the composite cathodes depends on the powder processing routes as well as on the coating technology (screen printing, slurry spraying) applied, thus, on the porous structure of electrode layers (thickness, porosity) [9]. A big influence of the sintering procedure (temperature, co-firing of cathodes, anodes, and electrolytes) has been established [9]. These results suggest that the interaction between the electrolyte (fluorite structure) and perovskite cathode, therefore, the properties of catalytically active interlayer, are very sensitive to the processing procedures and crucial for electrocatalytic activity of the SOFCs. Thus, the sintering temperature and durability of cathodes are the most important treatment parameters determining the electrochemical activity and kinetic characteristics (overvoltages, rate constants etc) of SOFCs.
Results of theoretical calculations show that the oxygen ion migration energy is determined by the transition state properties, i.e., by the so called “triangle” plane characteristics [10, 11]. One d-metal cation and two A-site cations compose the triangle for holding back the oxygen ion migration. Under the aggravating circumstances for the migration process, the bond between the transition metal cation and oxygen anion is not fully ionic, but has a slight covalent nature [10, 11]. Thus, the migration energy of oxygen ions depends on the ability of d-metal (Co, Fe, Ni, Mn) to change the oxidation state, because the partial charge transfer process occurs in the transition state [10–12]. Analysis of experimental results confirms that the redox behavior and characteristics of the B site cations have the key role in determining the LSC and LSCF electrocatalytic properties [13]. The flexible redox behavior of cobalt cationic centers provides the high oxygen flux at higher temperatures. However, it causes a large thermal expansion coefficient for Co cations containing perovskites, and the valence instability of cobalt cations introduces inherent phase instability of the LSC cathodes at intermediate temperatures (773–1073 K) [14].
Hjalmarsson et al. [8] analyzed the role of H2O in cathode gas for LSC cathode characteristics. They proposed the formation of Sr (OH)2 on the surface of the cathode. Moreover, it was expected that the hydroxide occupies the electrochemically active surface sites. Within the long lasting testing period, the strontium cation diffused from the cathode and the stoichiometric composition changed, having a significant influence on the chemical and electrochemical properties of SOFCs single cells. Also, it was mentioned that the oxygen vacancy can be blocked by hydroxide ions [8].
Ishihara et al. [15] showed some increase of activity against electrochemical oxygen reduction reaction in humidified air on Ba0.6La0.4CoO3−δ cathode. The improved properties of oxygen surface exchange for the mentioned cathode have been explained by accelerated catalytic oxygen dissociation in the presence of H2O.
In this work, the electrochemical in situ high-temperature X-ray diffraction (HT-XRD) studies have been conducted to characterize the changes in the phase composition of cathode electrode connected with transformations in cell volume, as well as in crystallite sizes. Electrochemical in situ XRD method, i.e., thermoelectrochemical expansivity study gives valuable opportunities to collect experimental data within the real working conditions and to analyze influence of the operating parameters like cell potential, temperature, and gas content in the cathode compartment on the crystallographic and electrochemical parameters of the electrode materials under study. Thus, changes in the lattice parameters of La0.6Sr0.4CoO3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ have been established at the various fixed temperatures, cathode potentials, and p O2 and p H2O conditions.
Experimental
Symmetrical cells for HT-XRD (773–1073 K) studies were prepared using gadolinia-doped ceria (Ce0.9Gd0.1O2-δ ) (GDC) electrolyte pellet as a support with thickness d el of nearly 750 μm [16–18]. The micro-mesoporous electrodes with geometrical flat-cross section surface area of 1.77 cm2 (both cathode and anode) were screen printed onto the GDC electrolyte. The LSC and LSCF powders with different stoichiometry were prepared by using the thermal combustion of corresponding nitrate solutions, where La(NO3)3∙6H2O, Sr(NO3)2 (all from Aldrich, 99.9 %), Co(NO3)2∙6H2O (98 %, Riedel de Haën), and Fe(NO3)3∙9H2O (Merck, 99 %) as the precursors, and glycine (99 %, Sigma-Aldrich) as the reducing agent were used [16–18]. Terpineol (Solsperse 3000), as dispersant, polyethylene glycol (as a binder), and polyvinyl butyral (as a plasticiser) were used for preparation of the viscous electrode pastes. The symmetrical electrodes deposited onto the GDC pellet were sintered at 1373 K for 5 h [16–18]. Electrical contacts were made using porous platinum contact layers, formed from Pt paste (MaTeck) during thermal treatment at T = 1273 K and connecting electrodes through the platinum wires to the Solartron measurement system. To control the potential of the electrode studied, the Pt/porous Pt in O2 (Pt/Pt/O2) reference electrode was prepared and fixed on the same side approximately 1 mm from the working electrode. The experiment was performed on the gas overflow conditions, where the oxygen and nitrogen gas flows were controlled by rotameters from Brooks Instruments. The symmetric single cells were investigated at fixed temperatures: 673, 773, 873, and 973 K and at different oxygen partial pressures: 0.03, 0.2, and 1 atm, applying the fixed electrode potentials. The H2O concentration was 1.7 wt% in synthetic air (21 % O2 + 79 % N2).
Detailed characterization of crystallographic structure of cathode has been performed by X-ray diffraction method applying a Bruker-AXS D8 instrument with a Cu Kα radiation source ((40 kV, 40 mA) a Goebel mirror, 2.5° Soller slits, and LynxEye 1D detector). An X-ray thermo-diffractometry study has been conducted using an Anton Paar HTK 1200 N temperature vessel. The SOFC single cell was placed into an alumina sample holder which was heated in different oxygen concentrations from room temperature (RT) up to 1073 K. For the XRD studies, a 60 min continuous scan from 21 to 75° 2Θ with 0.015° 2Θ step, and with total count time of 166 s per step, was applied. The detailed XRD single peak scan experiments were performed from 57.5 to 59.5° with 0.015° 2Θ step, and with a 40 s total count time per cycle.
The cell potential was controlled and electrochemical measurements were performed using the Solartron 1287 A potentiostat/galvanostat. The impedance spectra were recorded in a three electrode setup within the frequency range from 1 MHz to 0.01 Hz. The ac voltage amplitude was 5 mV. The Zview 2.2 software was applied for fitting the impedance data.
Results and discussion
The data in Fig. 1 show the micro–mesoporous cathode structure before and after the treatment with humidified (1.7 wt% of H2O) in synthetic air. As it can be seen from the figures, the H2O addition in oxidizing gas does not change the microstructure of the cathode. The high conductivity values for different single cells, obtained from the very high ac frequency impedance spectroscopy data (analyzed later), confirm that there is a very good contact between the electrolyte and cathode electrodes. The electrolyte layer appears almost as fully dense phase and contains only some isolated closed pores, but no pinholes or cracks have been observed. Macropores (pores wider than 50 nm) have been observed in a cathode microstructure. Based on the data in Refs. [16–20], the macropores act as transport channels and increase the gas permeability (oxidant and fuel diffusion) onto the reaction sites.

SEM images (15 kV, secondary electron detector) for cathode cross-section interface of half cells La0.6Sr0.4CoO3−δ a at the beginning and b after synthetic air +1.7 wt% H2O vapor treatment during 300 h. Sintering temperature of electrode was T Sint = 1323 K
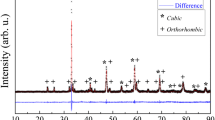
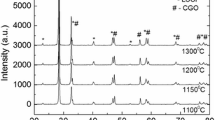
As it is demonstrated in Fig. 2, the 1.7 wt% H2O addition at T = 873 K for 100 h does not change the cathode crystal structure noticeably or the changes take place within the very thin surface layer of electrode, being not detectable by XRD method applied for signals over 100 h. The characteristic XRD patterns for the studied phases were confirmed via profile fitting method applying the Topas 4 software suite [21]. The XRD peaks for the studied LSC and LSCF materials are quite narrow and well expressed, and respond to 70 to 300 nm size crystallites calculated according Scherrer equation. Somewhat wider peaks can be seen for the A position rich (La0.6Sr0.4)1.01CoO3−δ cathode. The particles of electrode material (with diameter of about 2 μm) are composed of smaller (disoriented) particles with the crystallite sizes from 70 nm to 300 nm, obtained using the XRD data and Scherrer calculation method. However, the intensity of peaks is somewhat different from the corresponding reference peaks, given in the database (ICSD collection codes no. 86,944, 86,123, 160,393, and 82,817 [22–25]), due to the disoriented structure of La0.6Sr0.4CoO3−δ . The crystallite sizes are depending on the cathode synthesis conditions [16–18]. Based on the XRD data, given in Figs. 1 and 2, there are no traces of other chemical phases, like CoFe2O4 (which can be formed under electric polarization conditions) or some carbonates (synthesis products from raw powder). Thus, the perovskite phase with rhombohedral space R-3cR group structure has been detected for all the (La0.6Sr0.4) y CoO3−δ electrodes (0.99 ≤ y ≤ 1.01) under study [26, 27].

Selected segment from XRD pattern for the LSC cathode in dry synthetic air and in humidified synthetic air (H2O = 1.7 wt% in air) at different working times, noted in figure
More amorphous structure has been detected for the (La0.6Sr0.4)1.01CoO3−δ cathode, compared with the other studied (La0.6Sr0.4)0.99CoO3−δ and La0.6Sr0.4CoO3−δ electrodes (Fig. 3). It was observed that a slight deficiency of cations in A site increases the stability of perovskite structure. In addition, the XRD data indicate that the more amorphous (La0.6Sr0.4)1.01CoO3−δ cathode is less stable in humidified synthetic air at higher working temperatures during the long lasting testing period.

Segment from XRD pattern for the LSC cathodes with different A position composition, after treatment in humidified synthetic air for 10 days at T = 923 K, measured at room temperature
The total polarization resistance values, R p, calculated from the Nyquist plots [14–18] (Figs. 4–6) indicate that the electrochemical activity is lower for the (La0.6Sr0.4)1.01CoO3−δ cathode if compared with (La0.6Sr0.4)0.99CoO3−δ , and this material was more sensitive to humidified gas feeding parameters. The data in Figs. 4–7 show that the (La0.6Sr0.4)0.99CoO3−δ and La0.6Sr0.4CoO3−δ cathodes are more active against oxygen reaction process than the (La0.6Sr0.4)1.01CoO3−δ and La0.6Sr0.4Fe0.8Co0.2O3−δ electrodes.

Nyquist plots measured under various gas feeding conditions (1) at the beginning of test applying only the dry air, (2) after 10 days testing in dry air, and (3) after 10 days testing in the hymidified synthetic air
The Nyquist plots, given in Fig. 4 show that after 10 day testing in dry synthetic air, the increase of R p was only about 10 %. (curve 2). Thus, the degradation of cathode was minor. However, after 10 days testing in humidified synthetic air, as clearly demonstrated in Fig. 4 (curve 3), the degradation of cathode was much bigger (about 270 %). It is interesting that the very high frequency (f > 100 Hz) series resistance R ex = Z´ (f → ∞), calculated from complex impedance plane plot, does not change, however, the total polarization resistance R p increased remarkably. It is probably caused by the formation of Sr(OH)2 at the cathode surface, thus, by replacement of O2− vacancies by OH− ions. As the Sr(OH)2 formation takes place on/in the very thin upper layer of porous cathode surface, the XRD method cannot detect some possible changes in the La0.6Sr0.4CoO3−δ cathode surface structure as there is no big changes within all cathode layer. Surprisingly, after starting feeding of the dry synthetic air into the cathode compartment, (i.e., changing gas phase composition back to the dry air conditions), the polarization resistance increases continuously, demonstrated in Figs. 5 and 6. It must be noted that for all the La0.6Sr0.4CoO3−δ cathodes tested, including the (La0.6Sr0.4)1.01CoO3−δ electrode, the polarization resistance does not decrease after switching back to the dry gas feeding conditions, differently from La0.6Sr0.4Fe0.8Co0.2O3−δ (Fig. 7), where remarkable decrease of R p has been measured after switching gas phase back to the dry synthetic air.

Nyquist plots for the La0.6Sr0.4CoO3−δ measured under various gas feed conditions: (1) at the beginning of test in dry air, (2) after testing in humidified air during 100 h, and (3) in dry synthetic air after 100 h polarization in moistured air and thereafter testing for 300 h in dry synthetic air

Nyquist plots for the (La0.6Sr0.4)0.99CoO3−δ measured at T = 923 K, ΔE = −0.1 V and under various gas feeding conditions: (1) at the beginnig of test in dry air, (2) after testing in humidified air during 100 h, (3) after treatment in humidified synthetic air for 250 h, and (4) after treatment in humidified synthetic air for 320 h

Nyquist plots for the La0.6Sr0.4Fe0.8Co0.2O3−δ measured under various gas feed conditions (1) at the beginnig of test in dry air, (2) after 80 h in humidified air and (3) after testing for 80 h in humidified and thereafter 100 h in dry air
At higher temperatures (T ≥ 900 K), there are some changes in the Nyquist plots in very low frequency region (f < 1 Hz) (Fig. 6). This Nyquist plot region has been connected with the gas phase transport limitations in the macroporous cathode structure. Thus, the so called gas phase mass transfer resistance R mass, increases noticeably during 320 h testing time (compared with the beginning of experiment). The same tendency has been established for the La0.6Sr0.4CoO3−δ and (La0.6Sr0.4)1.01CoO3−δ cathodes. After turning feeding of the humidified synthetic air back to the dry air, the polarization resistance increases continuously (similarly to lower temperatures).
However, somewhat different behavior has been observed for the La0.6Sr0.4Fe0.8Co0.2O3−δ cathode (Fig. 7). Some increase of R p has been established after feeding of 1.7 wt% humidified air into the cathode compartment (R p increases ~250 % during 80 h feeding of 1.7 wt% + synthetic air). After feeding back of dry synthetic air, the total polarization resistance started to decrease, and after 100 h dry air feeding, the ΔR p = 215 %, if compared with the starting R p values.
The OH− anion formation kinetics depends on the B-site cation ability to change the oxidation state. Based on the data in literature [8] and our experimental results, the formation of OH− takes place mainly on the B-sites of perovskite cathodes. As the cobalt ion changes the oxidation state more easily compared with iron cation, the amount of O−2 ions decreased more quickly for the LSC cathodes. As there are OH− ions in the lattice, i.e., in the cathode upper layers, the lattice is more inflexible because Co4+ cations have been reduced into Co3+ states and the O−2 diffusion (mass transfer) is paralyzed. It should be mentioned that, the high frequency area of Nyquist plots characterizing the rate of faradic charge transfer processes does not change, and the charge transfer process also does not change. This phenomenon refers that the mass transfer processes are limiting for the oxygen reduction process on/in-side the porous cathode structure of studied materials.
The rate of oxygen exchange reaction on the cathode surface is quite high compared with the other processes taking place (including mass transfer processes: different diffusion and migration processes) on/in the porous electrode surface. For the LSCF cathode, the OH− anion formation/adsorption seems to be more reversible and takes places in the very thin upper layer of cathode, probably, contrary to the LSC cathodes with different A position concentration, and only Co cations in B position were the OH− anions formation/adsorption, seems to be irreversible.
From the Bode phase angle vs log f plots (Fig. 8), we can see that the characteristic time constants for the rate limiting processes are somewhat different for the LSCF cathode compared with the LSC cathodes. The exchange reaction for ½ O2 + 2e−→ O−2 is quite intensive and quick for LSC as well as LSCF materials within the high frequency area (f > 100 Hz), and the absolute phase angle values are very small │Θ│Θ < 5. This is in a good accordance with the fact that the oxidation state of Co cation in B-site is more flexible, if compared with that for Fe3+ cation. There is mixed kinetic limitation behavior for all the studied materials within moderate frequency region from 500 to 0.1 Hz, but the balance, between the rate limiting steps is shifted toward charge transfer limitation step for the LSCF cathode at higher f in an agreement with higher frequency values at the position of phase angle minimum.

Bode, phase angle vs log f plots for the La0.6Sr0.4Fe0.8Co0.2O3−δ (LSCF, open symbols), and for the La0.6Sr0.4CoO3−δ (LSC, filled symbols), measured at the beginning of experiments in dry air at different working temperatures, noted in figure
The rate limiting processes rates depend in addition to the chemical composition on the temperature applied. At low working temperatures (T < 873 K) for all materials, the under study, the phase angle absolute values are somewhat higher (from −10 to −20 degree) being characteristic of the mixed kinetics (mass transfer and charge transfer steps) if compared with T ≥ 923 K. Thus, for the La0.6Sr0.4Fe0.8Co0.2O3−δ cathode, at lower T ≤ 823 K, there are two mixed kinetic processes within the frequency region from 1000 to 1 Hz. With the increase of T, the rate of slower process increases and at T ≥ 973 K, there seems to be only one rate determining process characterized with a frequency minimum (f min) from 30 to 60 Hz. For La0.6Sr0.4CoO3−δ , f min values are two orders lower and demonstrating that the characteristic relaxation times for faradic charge transfer processes are slower.
It is surprizing, that for more active La0.6Sr0.4CoO3−δ cathode, some new slow processes can be seen at T ≥ 873 K explained by the slow mass transfer of H2O or O2 in the microporous structure of cathode (Fig. 9).

Bode plots for the (La0.6Sr0.4)0.99CoO3−δ measured at T = 923 K, ΔE = −0.1 V and under various gas feeding conditions: (1) at the beginning of test in dry air, 2) after testing in humidified air during 100 h, (3) after treatment in humidified synthetic air for 250 h, and (4) after treatment in humidified synthetic air for 320 h
Fitting of Nyquist plots
For the more detailed analysis of processes inside the cathodes, the complex impedance plane plots, calculated applying the equivalent circuit given in Fig. 10, have been fitted to the experimental Nyquist plots applying the least squares fitting method. The data established are given in Figs. 11 and 12. We can see that the series high frequency resistance (R 2), medium frequency resistance (R 3), and low frequency resistance (R 4) values depend on the electrode potential and temperature applied. There is a very well expressed dependence of mentioned parameters on the polarization time under humidified synthetic air feeding conditions. It is interesting that at fixed T = 923 K, the high frequency series resistance R 2 decreases with the polarization time applied in humidified cathode gas feeding conditions, indicating degradation of porous structure. The medium frequency R 3 values, similarly to R 4 values, increase with the polarization time in 1.7 % H2O + synthetic air environment. However, differently from R 3 vs ΔE – plots, where R 3 decreases with the rise of negative cathode potential, R 4 values are practically independent of ΔE, indicating that the rate of mass transfer (finite length diffusion) processes in microporous matrix is independent of ΔE applied. However, R 4 values noticeably increase with the polarization time applied, demonstrating blocking of smaller pores.

Equivalent circuit used for modeling of the experimental impedance spectra. R 1 is very high frequency series resistance, R 2 is high-frequency charge transfer resistance, R 3 is medium-frequency charge transfer resistance and R 4 is low-frequency charge transfer resistance. C 1, CPE 1 and CPE 2 are capacitance and constant phase elements, respectively

Equivalent circuit components R 2 (a), R 3 (b), and R 4 (c) dependences on ΔE: (1) at the beginnig of test in dry air, (2) after treatment in humidified synthetic air for 100 h, (3) after treatment in humidified synthetic air for 250 h, (4) after treatment in humidified synthetic air for 320 h

Equivalent circuit components R 2 (a), R 3 (b), and R 4 (c) vs ΔE dependences at the beginning of test in dry air conditions, at different temperatures, noted in figure
As mentioned earlier, the OH− ion formation/adsorption seems to be irreversible for LSC material. As we can see from Table 1, the R 3 and R 4 values increase for both materials (LSC and LSCF) in humidified conditions. After change dry synthetic air, the R 3 values start to decrease for LSCF material, contrary to the LSC cathode.
At the fixed polarization times and cell potential, R 2, R 3 values noticeably decrease if T ≤ 923 K. At T ≥ 923 K, R 2, R3, and R 4 values are very low and only weakly decrease with the increase of T. Interestingly, at higher T ≥ 873 K, R 2, R 3, and R 4 values only weakly depend on the partial oxygen pressure, if p O2 ≥ 0.20 atm, explained by the saturation of surface layer by O2 adsorption and intermediates of disproportion processes (Fig. 13).

Equivalent circuit components R 2 and R 4 dependences at the beginning of test in dry air conditions, at different oxygen partial pressures, noted in figure
Conclusion
The H2O addition in synthetic air does not change the crystal phase structure of cathode or the changes that take place within very thin surface layers of electrodes, however, not detectable using HT-electrochemical XRD method. In humidified synthetic air, the OH− anion adsorption and formation of Sr(OH)2 on the LSCF cathode are nearly reversible and take places in very thin upper layer of cathode, probably, contrary to the LSC cathode, where the OH− formation is irreversible. More amorphous structure has been detected for the (La0.6Sr0.4)1.01CoO3−δ cathode, compared with that established for the other (La0.6Sr0.4)0.99CoO3−δ and La0.6Sr0.4CoO3−δ cathode electrodes studied. It was observed that a small deficiency in A site increases the stability of perovskite structure.
The limiting process rates are somewhat different for the LSCF cathode as compared with the LSC cathodes. There are mixed kinetic limitation for both studied materials. Within the frequency region from 1000 to 0.1 Hz, the balance between rate limiting steps is shifted to near charge transfer limitation for the LSCF and LSC cathodes with the increase of T. The characteristic frequency values, i.e., characteristic time constants are smaller for the LSCF cathode than that for the LSC cathodes, indicating that only upper surface layer is catalytically active for the LSCF cathode, in agreement with the total polarization resistance values and charge transfer resistance values established using the non-linear least-squares fitting method of Nyquist plots.
References
Singhal SC, Kendall R (2003) High temperature solid oxide fuel cells: fundamental, design and application. Elsevier, Amsterdam
Minh NQ, Takahashi T (2005) Science and Technology of Ceramic Fuel Cells. Elsevier, Amsterdam
Wachsman ED, Singhal SC (2009) The electrochemical society. Interface 18:38–43
Zhao F, Virkar A (2005) J Power Sources 141:79–95
Bidrawn F, Kim G, Aramrueang N, Vohs JM, Gorte RJ (2010) J Power Sources 195:720–728
Steele BCH (2000) Solid State Ionics 129:95–110
Meadower DB (1970) Nature 226:847–848
Hjalmarsson P, Søgaard M, Mogensen M (2008) Solid State Ionics 179:1422–1426
Park HC, Virkar AV (2009) J Power Sources 186:133–137
Kotomin EA, Mastrikov YA, Kuklja MM, Merkle R, Roytburd A , Maier J (2011) Solid State Ionics 188:1–5
Mastrikov YA, Merkle R, Kotomin EA, Kuklja MM, Maier J (2013) Phys Chem Chem Phys 15:911–918
Hagen A, Traulsen ML, Kiebach WR, Johansen BS (2012) J Synchrotron Rad 19:400–407
Feldhoff A, Martynczuk J, Arnold M, Myndyk M, Bergmann I, Šepelák V, Gruner W, Vogt U, Hähnel A, Woltersdorf J (2009) J Solid State Chemistry 182:2961–2971
Kivi I, Aruväli J, Kirsimäe K, Heinsaar A, Nurk G, Lust E (2015) J Electrochem Soc 162:F354–F358
Ishihara T, Fukui S, Nishiguchi H, Takita Y (2003) ECS Trans 7:601–610
Lust E, Küngas R, Kivi I, Kurig H, Möller P, Anderson E, Tamm K, Samussenko A, Lust K, Nurk G (2010) Electrohim Acta 55:7669–7678
Küngas R, Kivi I, Lust E (2009) J Electrochem Soc 156:B345–B352
Küngas R, Kivi I, Lust K, Nurk G, Lust E (2009) J Electroanal Chem 629:94–101
Singhal SC (2008) Ind Ceram 28:53–57
Küngas R, Bidrawn F, Mahmoud E, Vohs JM, Gorte RJ (2012) Solid State Ionics 225:146–150
Kivi I, Aruväli J, Kirsimäe K, Heinsaar A, Nurk G, Lust E (2013) J Electrochem Soc 160:F1022–F1026
Sathe VG, Pimpale AV, Siruguri V, Paranjpe SK (1998) J. Phys. Condens Matter 10:4045–4055
Sonntag R, Neov S, Kozhukarov V, Neov D (1997) J.E. Elshof. Phys B Condens Matter 241:393–396
Šwierczek K, Gozu M (2007) J Power Sources 173:695–699
Hjalmarsson P, Søgaard M, Hagen A, Mogensen M (2008) Solid State Ionics 179:636–646
Sathe VG, Pimpale AV, Siruguri V, Paranjpe SK (1996) J. Phys. Condens Matter 8:3889–3896
Masatomo Y, Taisuke K (2008) Solid State Ionics 178:1939–1943
Acknowledgments
This research was supported by the EU through the European Regional Development Fund (TK141 “Advanced materials and high-technology devices for energy recuperation systems”), Estonian Science Foundation ETF grant no. 9352, the Estonian Centre of Excellence in Science Project TK 117 T, the Estonian Energy Technology Progam project SLOKT10209T, and European Spallation Source: Estonian Partition in ESS Instrument design, development and building, and application for scientific research SLOKT12026T.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kivi, I., Aruväli, J., Kirsimäe, K. et al. Influence of humidified synthetic air feeding conditions on the stoichiometry of (La1-xSrx)yCoO3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ cathodes under applied potential measured by electrochemical in situ high-temperature XRD method. J Solid State Electrochem 21, 361–369 (2017). https://doi.org/10.1007/s10008-016-3379-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-016-3379-0