
Abstract
The interaction between SARS-CoV-2 Spike protein and angiotensin-converting enzyme 2 (ACE2) is essential to viral attachment and the subsequent fusion process. Interfering with this event represents an attractive avenue for the development of therapeutics and vaccine development. Here, a hybrid approach of ligand- and structure-based virtual screening techniques were employed to disclose similar analogues of a reported antiviral phytochemical, glycyrrhizin, targeting the blockade of ACE2 interaction with the SARS-CoV-2 Spike. A ligand-based similarity search using a stringent cut-off revealed 40 FDA-approved compounds in DrugBank. These filtered hits were screened against ACE2 using a blind docking approach to determine the natural binding tendency of the compounds with ACE2. Three compounds, deslanoside, digitoxin, and digoxin, were reported to show strong binding with ACE2. These compounds bind at the H1-H2 binding pocket, in a manner similar to that of glycyrrhizin which was used as a control. To achieve consistency in the docking results, docking calculations were performed via two sets of docking software that predicted binding energy as ACE2-Deslanoside (AutoDock, −10.3 kcal/mol and DockThor, −9.53 kcal/mol), ACE2-Digitoxin (AutoDock, −10.6 kcal/mol and DockThor, −8.84 kcal/mol), and ACE2-Digoxin (AutoDock, −10.6 kcal/mol and DockThor, −8.81 kcal/mol). The docking results were validated by running molecular simulations in aqueous solution that demonstrated the stability of ACE2 with no major conformational changes in the ligand original binding mode (~ 2 Å average RMSD). Binding interactions remained quite stable with an increased potential for getting stronger as the simulation proceeded. MMGB/PBSA binding free energies were also estimated and these supported the high stability of the complexes compared to the control (~ −50 kcal/mol net MMGB/PBSA binding energy versus ~ −30 kcal/mol). Collectively, the data demonstrated that the compounds shortlisted in this study might be subjected to experimental evaluation to uncover their real blockade capacity of SARS-CoV-2 host ACE2 receptor.
Graphical abstract

Similar content being viewed by others

Introduction
In December 2019, a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has emerged in Wuhan Hubei province of China [1,2,3,4,5]. It has been identified as a previously unknown SARS-CoV that has crossed the species barrier to be known as SARS-CoV-2 and causes an atypical pneumonia [6]. This disease is now widespread and is known as coronavirus disease 19 or COVID-19. COVID-19 has turned into a pandemic and has spread across 210 countries across the globe, despite the efforts of health officials in China to limit its spread. It was declared as a pandemic by the WHO on 11th March 2020 [5]. The number of new cases and deaths are increasing with each passing moment. Betacoronavirus is recognized as the family and genus of SARS-CoV-2 which is known for highly pathogenic emerging viruses [7, 8]. The members of this family have been known for their lethality in both birds and mammals. Research is still being carried out on the two very famous viruses of this family that have caused hundreds of deaths over a period of 2 decades: severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) [9]. However, to date, scientists have been unable to find an effective treatment against this family of viruses. Currently, lots of therapeutics are under examination including: remdesivir, lopinavir with ritonavir, lopinavir with ritonavir plus interferon beta-1a, and chloroquine or hydroxychloroquine [10]. Therefore, this area is very open for research and study for the establishment of successful treatment methods to limit the spread of COVID-19.
For pursuing any study on SARS-COV-2, it is necessary to understand the structural units of a coronavirus. The basic structure of coronavirus includes an envelope [11, 12], membrane [13], nucleocapsid [14], and Spike proteins [15,16,17,18]. These are actually non-segmented positive sense RNA viruses [19]. Spike protein plays a vital role in attachment, fusion, and entry of virus into the host machinery, and hence, it is a main determinant of CoV tropism [17, 20,21,22]. It forms homotrimers that protrude from the surface of the virus and supports virus entry. These protrusions form spikes on the surface of virus giving it a crown-like appearance. In the initial phase of infection by the virus, angiotensin-converting enzyme 2 (ACE2) mediates the entry by fusion of viral and cellular membranes [17, 23]. This facilitates the binding of the Spike protein to the membrane receptor on the host cells. In case of coronavirus SARS-CoV-2 infection, human ACE2 is exploited for host cell entry [6, 24]. A recent study suggests another route of viral attack through CD147/S-protein [25]. Spike protein has two sub-units: S1 and S2 subunits. S1 binds to ACE2 through the receptor-binding domain (RBD); in this way, it mediates viral attachment to the cellular surface and hence entry into the host [26]. Viral entry also needs priming of Spike protein with serine protease TMPRSS2 in the host cell. This is followed by cleavage of Spike protein at sites S1/S2 and S2, which allow fusion of viral and cellular membranes. The S2 subunit drives the process of fusion [17]. Specific to SARS-CoV-2, a furin cleavage site is present at subunits S1 and S2 boundaries. The unique feature of SARS-CoV-2 from other coronaviruses is processing of the furin cleavage site during biosynthesis [27].
Human ACE2 is a type 1 membrane protein expressed in the heart, lung, intestine, and kidneys [28, 29]. It is the main target for viral entry by coronaviruses. The structure of ACE2 contains an N-terminal peptidase domain, a ~40-residue intracellular segment and a C-terminal collectrin-like domain that ends with a single transmembrane helix [30, 31]. A zinc-dependent PD domain is its single transmembrane helix which makes it daunting to establish the full length protein structure. This is the reason for limited availability for structural information regarding ACE2. Physiologically, it is a part of the renin-angiotensin system (RAS) and catalyzes cleavage of angiotensin II to angiotensin 1–7. The latter is a potent vasodilator in the cardiovascular system with defensive properties [32].
Recently, determination of a cryo-electron microscopy structure at a resolution of 3.5 Å of SARS-CoV-2 Spike trimer in prefusion conformation has been performed to facilitate countermeasure developments [21]. It was revealed that in this structure, one of the three RBDs rotated up to facilitate availability to the receptor. Biophysical and structural evidence is provided that shows binding of the SARS-CoV-2 protein ectodomain with PD of human ACE2 with an affinity ~10 to 20 fold higher than that shown in binding with SARS-CoV [21, 33]. The recognition of the viral RBD is mainly performed through polar residues of extracellular PD of ACE2. It is revealed in molecular studies that the two S protein trimers bind to an ACE2 homodimer simultaneously [33]. The residues in SARS-CoV-2 RBD critical for binding with ACE2 are also validated through X-ray crystallographic data. The majority of these residues are highly conserved and the side chain properties are similar for both SARS-CoV-2 and SARS-CoV RBDs [34]. Largely, the COVID-19 research is being focused on production of vaccines [35] and targeting of nonstructural proteins: main protease (Mpro) and RNA-dependent RNA polymerase (RdRp) [6]. However, our focus is on investigation of novel infection prevention strategies against SARS-CoV-2. In 2004, crystal structures of free and bound forms of ACE2 were first solved at the resolutions of 2.2 Å and 3.0 Å, respectively [36]. Huentelman et al. discovered a novel inhibitor against ACE2 using structure-based virtual screening approach for a chemical library of ~140,000 diverse synthetic compounds [37]. The novel compound proved effective in blocking the S protein-mediated SARS-CoV cell fusion. The findings of this study support drug repositioning as a concept. Drug repositioning is also known as drug repurposing and it describes the process of creating novel clinical opportunities for available drugs [38]. This process, in comparison to de novo drug discovery, truncates the chemical structure optimization time and reduces the toxicological testing time. Previously for the treatment of other coronavirus infections, including MERS-CoV, this strategy has also been used [39]. In the current study, employing a molecular mechanics (MM)-assisted structure-based virtual screening method, an investigation of glycyrrhizin-based inhibitors to act as potential inhibitors of ACE2 has been undertaken. Glycyrrhizin is a plant-based derivative and has been reported to show anti-HVC, anti-HIV, and anti-ACE activity [40, 41]. This compound has been demonstrated to effectively neutralize SARS-CoV-2 in vitro and bind to the ACE2 receptor [40, 42,43,44,45,46]. New drug molecules are uncovered in this work, which are structurally related to glycyrrhizin and have been already approved by the Food and Drug Administration (FDA). Thus, the screened drugs can be repurposed against COVID-19 pandemic disease.
Materials and methods
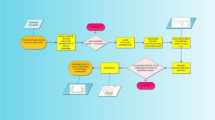
Figure 1 demonstrates the methodology of this work.

Schematic representation of the flow of steps used in the present study
Retrieval of ACE2 receptor structure and processing
The 3D structure of the ACE2 receptor was retrieved from RCSB Protein Databank (PDB ID: 6LZG) [34]. The initial PDB file was treated in a preparatory phase where all non-relevant co-crystalized structures and ligands were discarded in UCSF Chimera, alpha version 1.15 [47]. A short energy minimization run was then performed to add missing atoms if left in the crystallization process and remove steric clashes. During energy minimization, the first hydrogen atoms were added to the protein and missing bonds were repaired. The structure was then subject to 100 steps of energy minimization via steepest descent and 10 steps of conjugate gradient algorithms while keeping the step size to 0.02 Å.
Ligand based similarity search
Glycyrrhizin, which is a phytochemical and has been extensively documented for its potential for antiviral activity as well for its ability to block SARS-CoV ACE2 receptor, was used as the parent compound in the ligand-based virtual screening [40, 41, 48]. The similarity search was done in DrugBank 5.0 [49] employing a very stringent threshold of 0.6 (60%) and FDA-approved drugs. All the structures of the potent compounds were extracted from DrugBank in MDL Mol format and converted into PDB format using Open Babel [50]. Pre-optimization was performed with Chem3D Pro software [51] employing an MM2 force field [52]. The compounds were then uploaded to PyRx 0.8 [53] where they were converted to pdbqt. Meanwhile, the ACE2 receptor was also added to the PyRx AutoDock panel in preparation for structure-based virtual screening [54].
Molecular docking
For molecular docking and scoring procedures, PyRx AutoDock 4.2.6 was used [55]. To allow free binding of the compounds with ACE2, a blind docking strategy was applied where the search space center at XYZ coordinates was set as −25.16 Å, 15.93 Å, and − 25.12 Å, respectively. The grid dimensions were as follow: X (62.34 Å), Y (69.55 Å), and Z (71.84 Å). The number of iterations set for each compound was 100. Also, the criterion for RMSD values and binding energy values in docking experiments was around 2 Å and below −7 kcal/mol, respectively. The selected compounds were subjected to PreADMET toxicity predictor for Ames mutagenicity and rat carcinogenicity evaluation [56].
Molecular dynamics simulations
Molecular dynamic studies were carried out to evaluate the binding force of compounds in the active site, as shown by the docking studies. The role of water molecules and conformational dynamics of the ACE2 were also assessed. A total of 50 ns full-scale atomic simulations were undertaken using AMBER18 software [57] with a general atom force field (GAFF) [58] for small molecules and ff14SB [59] for ACE2. The antechamber in AMBER [60] was used to add atomic charges, together with a force field for the purpose of generating parameters of compounds for non-bonded interaction with enzymes using the AM1-BCC method [61]. All the atoms and bonds were incorporated according to their topologies after analysis of the structures. Leap (tleap) was used to build topologies and initial coordinates of complexes to run the molecular dynamics simulations [62]. Simulations were performed under periodic boundary conditions in a rectangular size cell with dimensions 65 × 65 × 75 Å3 with a 10 Å non-bonded cut-off and using Particle Mesh Ewald (PME) for the long-range electrostatics [63]. Initially, ACE2-compound complexes were subjected to energy minimization using a total of 1000 minimization steps with 500 steepest descent and 500 conjugate gradient steps to 0 K. In this way, temperature was raised to 100 K in the first cycle. Later, the system was equilibrated to 300 K in the next cycle. Finally, the production run was carried out at a constant temperature and pressure for 50 ns each with an integration step size of 2 fs using SHAKE [64]. Simulations were run at a pH value of 7 and the protonation state of all residues was kept fixed. This was done according to the fact that the virus interactions with the ACE2 are activated at neutral pH [65]. System stability was examined by calculating root mean square deviation (RMSD) for both ACE2 and compounds of all four complexes using AMBER CPPPTRAJ [66]. Additionally, hydrogen bond analysis for all frames of the simulation trajectories was completed using VMD [67].
Binding free energy calculation (MMGB/PBSA)
In this study, MMPBSA implemented in MMPBSA.py script was used for the binding free energy calculations [68, 69]. For this purpose, complexes were selected on the basis of best docking scores. In the first step, multiple snapshots (100) are generated from a molecular dynamics simulation production run trajectory for the respective complex. The final step is to calculate free energy for each snapshot and the formula for estimation of free energy is stated as:
Effective binding free energy is considered an entropic term and is ignored in the binding free energy calculation. In this way, energy calculations were performed for each complex.
Results and discussion
Glycyrrhizin, a triterpene glycoside, is a promising and safe antiviral natural product extracted from the roots of Glycyrrhiza glabra [40, 41, 48, 70]. This drug has a long established history of medical use and has been in use for treating atopic dermatitis, viral hepatitis, influenza A virus, Epstein Barr virus, varicella-zoster virus, parainfluenza virus type 2, dengue virus, chikungunya virus, Semliki Forest viruses, human immunodeficiency virus (HIV), and SARS-CoV and many more make this drug as a good candidate either alone or in combination to be evaluated against SARS-CoV-2 [71]. Considering this, we performed a ligand-based similarity search that filtered 40 compounds as analogs of glycyrrhizin and used in screening against SARS-CoV-2 ACE2 receptor (Table 1).
Molecular docking studies
Molecular docking is crucial in forecasting the predominant binding mode of a ligand molecule to a biological macromolecule and determining binding affinity of the complex [72, 73]. Traditionally, this process is tedious, long, and costly. The introduction of computer-aided drug design undoubtedly played a significant contribution in overcoming the many limitations of conventional drug design and rationalizing the drug discovery process [74]. Molecular docking was swiftly found to have value in the modern structure-based drug design in predicting the binding mode and intermolecular framework of chemical interactions between small drug molecules and proteins that drive inhibition of protein functionality [75]. The reported antiviral agent glycyrrhizin and a library of its closed 40 analogues were screened against the ACE2 receptor. To ensure consistency in the docking process, we initially tested the docking protocol by running a blind docking calculation of the co-crystalized ligand with ACE2 receptor. An identical binding mode was predicted, thus affirming the accuracy of the docking process. Further, consistency in the compounds’ binding mode was examined by performing docking the compounds with ACE2 via two docking platforms. It was also ensured that selected compounds strongly bind to the ACE2 residues that are in direct contact with Spike protein [34]. The compounds used in the screening process were ranked based on the binding energy, and the results were compared to opt top 3 molecules that have the least binding energy in kcal/mol. The best binding complexes are ACE2-Deslanoside (AutoDock binding energy = −10.3 kcal/mol and DockThor = −9.53 kcal/mol), ACE2-Digitoxin (AutoDock binding energy = −10.6 kcal/mol and DockThor = −8.84 kcal/mol), and ACE2-Digoxin (AutoDock binding energy = −10.6 kcal/mol and DockThor = −8.81 kcal/mol). The control ACE2-Glycyrrhizin complex has AutoDock binding energy = −10.3 kcal/mol and DockThor binding energy of −9.53 kcal/mol. A general description of the binding energy of all 41 compounds used in both of the sets of docking software is provided in Fig.2. The obtained docking result demonstrated that the minimum binding energy in AutoDock is −11.2 kcal/mol, with a maximum of −6.1 kcal/mol and mean of −8.42 kcal/mol. In case of DockThor, the minimum, maximum, and mean binding energy values are −9.64 kcal/mol, −6.8 kcal/mol, and − 7.91 kcal/mol, respectively. Moreover, for the ease of the experimentalists, the dissociation constant (Ki) value was predicted for the compounds: deslanoside (2.13 μM), digitoxin (1.48 μM), digoxin (1.17 μM), and glycyrrhizin (2.87 μM). The compounds were further screened for toxicity moieties and predicted to be negative for the Ames mutagenicity assay, and rat carcinogenicity.

Distribution of binding energy of compounds in both AutoDock and DockThor
Deslanoside-ACE2 complex
Deslanoside (4-(12,14-dihydroxy-3-((4-hydroxy-5-((4-hydroxy-5-((4-hydroxy-6-methyl-5-((3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-pyran-2-yl)oxy)-6-methyltetrahydro-2H-pyran-2-yl)oxy)-6-methyltetrahydro-2H-pyran-2-yl)oxy)-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)furan-2(5H)-one) is docked along helical 1 and 2 RBD binding sites at the ACE2 receptor. The binding conformation is towards the H3 helical region where the chemical moiety 4-(3a,7-dihydroxy-7a-methyloctahydro-1H-inden-1-yl)furan-2(5H)-one interacts with the H3 helical region through balanced hydrogen and hydrophobic contacts. The 6-(hydroxymethyl)tetrahydro-2H-pyran-2,3,4,5-tetraol of the compound interacts through rich weak hydrophobic binding. The central backbone chemical moiety of 6-((4-hydroxy-2-methyl-6-((4a-methyldecahydronaphthalen-2-yl)oxy)tetrahydro-2H-pyran-3-yl)oxy)-2-methyltetrahydro-2H-pyran-3,4-diol exhibited more affinity towards the H1 helical site compared to terminal rings that favor binding H2 helix. The docked conformation of the deslanoside and ACE2 and its chemical interactions are illustrated in Fig.3.

3D and 2D illustration of deslanoside binding mode and chemical interactions at the SARS-CoV-2 RBD binding site of ACE2 receptor. A Docked binding mode of Deslanoside (yellow color stick) respect to surface ACE2 (the most hydrophobic are tan whereas polar residues are colored are medium purple). B Different molecular interactions of deslanoside (shown in ball and stick) involved in the binding of deslanoside with ACE2 residues
Digitoxin-ACE2 complex
Like deslanoside, digitoxin (4-(3-((5-((5-((4,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-4-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-4-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-14-hydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)furan-2(5H)-one) is also aligned along the long cylindrical pocket of H1 and H2 binding pocket. The compound binding is dominated by hydrophobic interactions along the length of interactions with the pocket residues. The major center of the compound’s structure was elevated whereas the terminal moieties were dipped. The 4-(3a-hydroxy-7a-methyloctahydro-1H-inden-1-yl)furan-2(5H)-one moiety of the compound was configured to the H3 helix and adjusted near to the H1 helix. The opposite 6-methyltetrahydro-2H-pyran-2,4,5-triol part of the compound lay perpendicular on the cavity center to facilitate interactions with key residues. The docked conformation of digitoxin and ACE2 and its chemical interactions are illustrated in Fig.4.

Binding mode and chemical interactions of SARS-CoV-2-ACE2 and digitoxin. A Docked binding mode of digitoxin (yellow color stick) and ACE2 (hydrophobicity surface). B van der Waals interactions of digitoxin (shown in ball and stick) involved in the binding with ACE2 residues
Digoxin-ACE2 complex
Among the hits, digoxin (4-(3-((5-((5-((4,5-dihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-4-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-4-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-12,14-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)furan-2(5H)-one) is positioned exactly at the center of the H1-H2 pocket. The bulk of the 4-(12,14-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)furan-2(5H)-one of the compound moiety associates with the H2 helix where its furan-2(5H)-one ring is pointed to the H helix. The region 6-((4-hydroxy-6-((4-hydroxy-2-methyltetrahydro-2H-pyran-3-yl)oxy)-2-methyltetrahydro-2H-pyran-3-yl)oxy)-2-methyltetrahydro-2H-pyran-3,4-diol is sandwiched between the H1 and H2 helix. Here, the 6-methyltetrahydro-2H-pyran-2,4,5-triol ring formed strong hydrogen bonding with Gln42 and Lys68 residues of the pocket. The docked conformation of digoxin and ACE2 and its chemical interactions are illustrated in Fig.5.

Binding mode and chemical interactions of SARS-CoV-2-ACE2 and digoxin. A Docked binding mode of digoxin (deep pink color stick) and ACE2 (dark cyan hydrophobicity surface). B Chemical interactions of digoxin (shown in ball and stick) involved in the binding with ACE2 residues
Glycyrrhizin-ACE2 complex
The control glycyrrhizin (6-((6-carboxy-2-((11-carboxy-4,4,6a,6b,8a,11,14b-heptamethyl-14-oxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,14,14a,14b-icosahydropicen-3-yl)oxy)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)oxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid), as demonstrated before, showed great binding affinity for the ACE2 receptor and showed multiple strong hydrogen and hydrophobic interactions. In particular, 6-((6-carboxy-2,4,5-trihydroxytetrahydro-2H-pyran-3-yl)oxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid that contributed highly to the strong interactions anchored the compound at the H1-H2 pocket base and was supported on both helices. This allowed the rest of 2,4a,6a,6b,9,9,12a-heptamethyl-13-oxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,11,12,12a,12b,13,14b-icosahydropicene-2-carboxylic acid to project parallel towards the H3 helix. The docked conformation of glycyrrhizin and ACE2 and its chemical interactions are illustrated in Fig.6.

Binding conformation and chemical interactions of SARS-CoV-2-ACE2 and glycyrrhizin. A Docked binding mode of Glycyrrhizin (dodger blue color stick) and ACE2 (hydrophobicity chartreuse surface). B Chemical interactions of glycyrrhizin (shown in ball and stick) involved in the binding with ACE2 residues
Molecular dynamics simulation
Molecular dynamics simulation is a generally used technique to evaluate the stability and dynamic behavior of a biomolecule or biomolecule/ligand complex under different sets of conditions [76]. An affirmation was done on the initial conformation of the hit compounds-ACE2 complexes obtained after docking in water solution for 50 ns by all atom MD simulations. Uniformity in complex dynamics was noticed as measured by RMSD (Fig.7A). The RMSD values of the ACE2 alpha-backbone carbon atoms in the complex of the four possible inhibitors were measured with respect to the original docked structure which are Deslanoside-ACE2 complex (maximum, 2.79 Å and mean 1.92 Å), Digitoxin-ACE2 complex (maximum, 2.79 Å and mean, 2.14 Å), Digoxin-ACE2 complex (maximum, 2.40 Å and mean, 1.95 Å), and Glycyrrhizin-ACE2 complex (maximum, 2.40 Å and mean 1.95 Å). As can be seen in Fig.7A, all the systems were well converged and upon visualization of the complexes in different time frames reflected stability of the attached compounds to the ACE2 despite few structural changes to get strength in interactions. The stability of complexes was further validated by performing compound RMSD calculations and these were determined as Deslanoside-ACE2 complex (maximum, 2.95 and mean, 1.28), Digitoxin-ACE2 complex (maximum, 3.89 Å and mean, 2.22 Å), Digoxin-ACE2 complex (maximum, 3.50 Å and mean, 1.99 Å), and Glycyrrhizin-ACE2 complex (maximum, 3.50 Å and mean, 1.99 Å). The ligands from all four systems remained in a stable binding and conformation state, with the exception of digitoxin which revealed binding pose variations, as depicted in Fig.7B. Hydrogen bonds played a key function in stabilizing ligand binding at the docked site. The total number of hydrogen bonds formed by ACE2 with the three candidate compounds is shown in Fig.7C. Throughout the simulation time, all four compounds formed hydrogen bonds up to 4 in number with ACE2 receptor. These bonding parameters suggested that all candidate compounds have the ability to bind the ACE2 efficiently and tightly. Thus, the relative stability of the complexes filtered in this study has been inferred in terms of dynamics.

Molecular dynamics simulation trajectories analysis. A ACE receptor RMSD versus time, B ligands RMSD versus time, and C number of hydrogen bonds in each simulation frame
Binding free energy calculations
The MMGB/PBSA based binding free energy and molecular interactions between the shortlisted compounds and the ACE2 receptor complex have been calculated. Both methods have shown formation of very stable complex interactions. Table 2 illustrates the description of the obtained binding free energies. The entropy term is omitted since, and in some situations, it can not be measured due to convergence problems. As seen in the table, the term electrostatic energy, being most favorable, dominates the total energy of binding. The complex formation contributes to favorable non-polar interactions as compared to the polar part of the solution-free energy through non-favorable contributions. However, the overall van der Waals contribution is still high. The combined average values for the gas phase binding free energy using both MMGB/PBSA methods were determined to be a significant contribution towards overall complex stabilization. The 100 snapshots from the resulting trajectories were averaged for the calculation of all the energy values. All the molecular interactions were driven by polar and non-polar components in a polar solvent, where non-polar interactions and their contributions to energy were more advantageous. In the hydrophobic regions, non-polar residues show a tendency to hide and dissipate water molecules at the binding site. In order to obtain reliable results from absolute binding free energies, convergence of average values determined by MMGB/PBSA must be achieved. Energy oscillations are mainly due to limited sample size, so longer timescales are needed for snapshot extraction.
Per residue energy decomposition
To better understand the atomistic interaction mechanism and binding pattern of the compounds with ACE2, the residue wise free energy contribution, to the overall binding free energy of the complex, was calculated using MMGB/PBSA methods. All these residues are classified hotspot amino acids which have shown a contribution of <− 1 kcal/mol and have played a significant role in stabilizing the docked complex (Table 3). The residues that together make high free energy contributions <− 1 kcal/mol in both calculations (PB/GB) are Thr9, Phe10, Lys13, Phe14, Glu17, Ala18, Asp20, Leu21, Gln24, Ala47, Lys50, Phe54, Glu57, Gln58, and Asn61. Therefore, the residues greatly add to the total free energy of the studied system. All these residues lie either within or in close proximity of the binding pocket of ACE2 and display a considerably low energy value of in the binding pocket, highlighting their importance. Aforementioned residues are involved in hydrogen and hydrophobic bond interactions with the compounds resulting in a network of chemical interactions around it. The absence of any residue among the residues listed above might ultimately result in the loss of catalytic activity. It was shown that all the hotspot residues were present in or near the protein’s active site. Additionally, the binding free energy was classified into its components such as the electrostatic, van der Waals, and solvation contribution to define the detailed contribution of each key residue. As demonstrated earlier, the total binding free energy is dominated by electrostatic term contributions, and supports the previous results of the energy calculation. The involvement of some of the binding residues in hydrogen bond interactions was not found but these residues showed favorable van der Waals and non-polar energies attributed to their van der Waals contacts with the compounds. Significant polar contributions of ≤ − 1 kcal/mol were observed by most of the active site residues while combining the results of both free energy methods. Phe10, Leu21, Phe54, and Asn61 were found to make considerable contributions in MMGB/PBASA in binding with all compounds.
Conclusions
In this work, ligand-based and structure-based virtual screening approaches were explored for identification of FDA-approved potent chemical compounds as ACE2 inhibitors from DrugBank. Initial sorting based on ligand-based similarity screening resulted in formation of a library of 40 compounds. Further, a blind docking technique was considered that led to the selection of three bioactive molecules (deslanoside, digitoxin, and digoxin) after comparative analysis with control (glycyrrhizin). These compounds bind directly to the site where SARS-CoV-2 Spike interacts, so it is highly likely that these compounds will interfere with the Spike-ACE2 interaction thus hindering the binding and fusion event in the life cycle of the virus. Extensive dynamics evaluation of the complexes was also performed in an aqueous environment that elucidated stable conformational stability of the ACE2 and of the compounds. Binding free energy estimation showed the compounds had better binding affinity compared with the control. In the interactions, Phe10, Leu21, Phe54, and Asn61 were found to contribute significantly in stabilizing the compounds at the docked site. Hence, this investigation indicated that the selected compounds could be subjected to further investigation as a novel set of leads for the rational drug designing of ACE2 inhibitors in COVID-19 therapy.
Data availability
All data generated or analyzed during this study are included in this published article.
Code availability
N/A
References
Li, H., Liu, S.-M., Yu, X.-H., Tang, S.L. and Tang, C.K.: Coronavirus disease 2019 (COVID-19): current status and future perspectives. Int. J. Antimicrob. Agents. 55(5), 105951 (2020)
Hirano, T., Murakami, M.: COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity 52(2), 731-733 (2020)
Chakraborty, I., Maity, P.: COVID-19 outbreak: Migration, effects on society, global environment and prevention. Sci. Total. Environ. 728, 138882 (2020)
Wang, L., Wang, Y., Ye D, Liu, Q.: Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents. 55(6), 105948 (2020)
Ghebreyesus TA (2020) WHODirector-General’s opening remarks at the media briefing on COVID-19-11 March 2020. World Health Organization. Downloaded fromhttps://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020. Accessed 21 June, 2021
Terali K, Baddal B, Gülcan HO (2020) Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: insights from a molecular mechanics-assisted structure-based virtual screening experiment. J Mol Graph Model 100:107697
Chan JFW, Lau SKP, To KKW et al (2015) Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522
Huang C, Wang Y, Li X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. https://doi.org/10.1016/S0140-6736(20)30183-5
Lu R, Zhao X, Li J et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574
Organization WH, others (2020) Solidarity” clinical trial for COVID-19 treatments. World Heal Organ (WHO) Situat reports Geneva WHO[Accessed 5 Apr 2020] Available from https//www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19
Gupta, M.K., Vemula, S., Donde, R., Gouda, G., Behera, L., Vadde, R.: In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 39(7), 2617-2627 (2020)
Zhang, Y.-Z., Holmes, E.C.: A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 181(2), 223-227 (2020)
Gil, C., Ginex, T., Maestro, I., Nozal, V., Barrado-Gil, L., Cuesta-Geijo, M.Á., Urquiza, J., Ramírez, D., Alonso, C., Campillo, N.E. and Martinez, A.: COVID-19: drug targets and potential treatments. J. Med. Chem. 63(21), 12359-12386 (2020)
Kang, S., Yang, M., Hong, Z., Zhang, L., Huang, Z., Chen, X., He, S., Zhou, Z., Zhou, Z., Chen, Q. and Yan, Y.: Crystal structure of SARS-CoV- 2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B. 10(7), 1228-1238 (2020)
Yuan Y, Cao D, Zhang Y et al (2017) Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8:15092
Yi, C., Sun, X., Ye, J., Ding, L., Liu, M., Yang, Z., Lu, X., Zhang, Y., Ma, L., Gu, W. and Qu, A.: Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 17(6), 621-630 (2020)
Walls, A.C., Park, Y.J., Tortorici, M.A., Wall, A., McGuire, A.T. and Veesler, D.: Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 181(2), 281-292 (2020)
Rota PA, Oberste MS, Monroe SS et al (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science (80- ) 300:1394–1399
Chen, Y., Liu, Q. and Guo, D.: Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92(4), 418-423 (2020)
Goo, J., Jeong, Y., Park, Y.S., Yang, E., Jung, D.I., Rho, S., Park, U., Sung, H., Park, P.G., Choi, J.A. and Seo, S.H.: Characterization of novel monoclonal antibodies against MERS-coronavirus spike protein. Virus Res. 278, 197863 (2020)
Wrapp D, Wang N, Corbett KS et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80- ) 367:1260–1263
Hulswit RJG, De Haan CAM, Bosch B-J (2016) Coronavirus spike protein and tropism changes. Advances in virus research. Elsevier, pp 29–57
Walls AC, Tortorici MA, Bosch B-J et al (2016) Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531:114–117
Zhou P, Yang X-L, Wang X-G et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273
Liu, C. and Zhu, D.: Cyclophilin A and CD147: novel therapeutic targets for the treatment of COVID-19. Medicine in Drug Discovery, p.100056 (2020)
Korber, B., Fischer, W., Gnanakaran, S.G., Yoon, H., Theiler, J., Abfalterer, W., Foley, B., Giorgi, E.E., Bhattacharya, T., Parker, M.D. and Partridge, D.G.: Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. BioRxiv. (2020)
Davidson AD, Williamson MK, Lewis S et al (2020) Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med 12:1–15
Wu, C., Zheng, S., Chen, Y. and Zheng, M.: Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCoV, in the nasal tissue. MedRxiv. (2020)
Zhang H, Kang Z, Gong H, et al (2020) The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. BioRxiv
Donoghue M, Hsieh F, Baronas E et al (2000) A novel angiotensin-converting enzyme--related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87:e1–e9
Zhang H, Wada J, Hida K et al (2001) Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem 276:17132–17139
Paul M, Poyan Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86:747–803
Yan R, Zhang Y, Li Y et al (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (80- ) 367:1444–1448
Wang, Q.H., Song, H. and Qi, J.X.: Structure of novel coronavirus spike receptor-binding domain complexed with its receptor ACE2. RCSB PDB. (2020)
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12:254
Towler P, Staker B, Prasad SG et al (2004) ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem 279:17996–18007
Huentelman MJ, Zubcevic J, Hernandez Prada JA et al (2004) Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension 44:903–906
Elmezayen, A.D., Al-Obaidi, A., Şahin, A.T. and Yelekçi, K.: Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 39(8), 2980-2992 (2020)
Dyall J, Coleman CM, Hart BJ et al (2014) Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 58:4885–4893
Cinatl J, Morgenstern B, Bauer G et al (2003) Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 361:2045–2046
Luo, P., Liu, D. and Li, J.: Pharmacological perspective: glycyrrhizin may be an efficacious therapeutic agent for COVID-19. Int. J. Antimicrob. Agents. 55(6), 105995 (2020)
Chen, H. and Du, Q.: Potential natural compounds for preventing SARS-CoV-2 (2019-nCoV) infection. Preprints Med. Pharmacol. 202001.0358.v2. (2020)
Murck H (2020) Symptomatic protective action of glycyrrhizin (Licorice) in Covid-19 infection? Front Immunol 11:1239
Luo P, Liu D, Li J (2020) Pharmacological perspective: glycyrrhizin may be an efficacious therapeutic agent for COVID-19. Int J Antimicrob Agents 55:105995
Zhang L, Liu Y (2020) Potential interventions for novel coronavirus in China: a systematic review. J Med Virol 92:479–490
Gomaa, A.A., Abdel-Wadood, Y.A.: The potential of glycyrrhizin and licorice extract in combating COVID-19 and associated conditions. Phytomedicine Plus. 100043 (2021)
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Chrzanowski, J., Chrzanowska, A., Graboń, W.: Glycyrrhizin: An old weapon against a novel coronavirus. Phytother. Res. 35(2), 629-636 (2021)
Wishart DS, Feunang YD, Guo AC et al (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082
O’Boyle NM, Banck M, James CA et al (2011) Open Babel: an open chemical toolbox. J Cheminform 3:33
Irwin, J.J.: Software Review: ChemOffice 2005 Pro by CambridgeSoft. (2005)
Halgren TA (1996) Merck molecular force field. J Comput Chem 17:490–519.
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Chemical biology. Springer, pp 243–250
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Vieira TF, Sousa SF (2019) Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl Sci 9:4538
Lee, S.K., Lee, I.H., Kim, H.J., Chang, G.S., Chung, J.E. and No, K.T.: The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR designing drugs and crop protectants: processes, problems and solutions, pp.418-20 (2003)
Case, D.A., Walker, R.C. Cheatham, T.E.: Amber 18, Univ. California, San Fr. (2018)
Wang J, Wolf RM, Caldwell JW et al (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Maier JA, Martinez C, Kasavajhala K et al (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713
Wang J, Wang W, Kollman PA, Case DA (2001) Antechamber: an accessory software package for molecular mechanical calculations. J Am Chem Soc 222:U403
Jakalian A, Bush BL, Jack DB, Bayly CI (2000) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem 21:132–146
Schafmeister C, Ross WS, Romanovski V (1995) The leap module of AMBER. Univ Calif
Petersen HG (1995) Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys 103:3668–3679
Kräutler V, Van Gunsteren WF, Hünenberger PH (2001) A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem 22:501–508
Dimitrov DS (2003) The secret life of ACE2 as a receptor for the SARS virus. Cell 115:652–653
Roe DR, Cheatham III TE (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9:3084–3095
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Miller BR, McGee TD, Swails JM et al (2012) MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput 8:3314–3321. https://doi.org/10.1021/ct300418h
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discovery 10:449–461. https://doi.org/10.1517/17460441.2015.1032936
Crance JM, Scaramozzino N, Jouan A, Garin D (2003) Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antivir Res 58:73–79
Bailly, C. Vergoten, G.: Glycyrrhizin: An alternative drug for the treatment of COVID-19 infection and the associated respiratory syndrome?. Pharmacol. Ther. p.107618 (2020)
Morris GM, Lim-Wilby M (2008) Molecular docking. Molecular modeling of proteins. Springer, pp 365–382
Meng X-Y, Zhang H-X, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7:146–157
Menchaca TM, Juárez-Portilla C, Zepeda RC (2020) Past, present, and future of molecular docking. Drug Discovery and Development-New Advances. IntechOpen
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20:13384–13421
Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. BMC Biol 9:71
Funding
Dr. Yasir Waheed is highly grateful to the Foundation University Islamabad, for granting financial assistance to accomplish this work.
Author information
Authors and Affiliations
Contributions
Sajjad Ahmad designed the research, performed virtual screening, analyzed molecular dynamics simulation trajectories, and binding free energy results and drafted the manuscript. Yasir Waheed supervised the study and reviewed and edited the manuscript. Asma Abro helps in drafting the manuscript and critical review the manuscript. Sumra Wajid Abbasi performed molecular dynamics simulations and binding free energy assays. Saba Ismail helps in simulation analysis. All authors have read and approved the final manuscript.
Sajjad Ahmad: data curation, visualization, investigation, and writing—original draft preparation. Yasir Waheed: conceptualization, methodology, supervision, writing—reviewing and editing. Asma Abro: software and writing—reviewing and editing. Sumra Wajid Abbasi: writing—original draft preparation. Saba Ismail: data curation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Glycyrrhizin is a safe and potent plant based antiviral phytochemical.
• Glycyrrhizin based similarity search revealed 40 FDA-approved compounds.
• Deslanoside, digitoxin, and digoxin are strong binders of SARS-CoV-2 ACE-2 receptor.
• Filtered leads are stable dynamically with multiple chemical interactions to ACE2.
• The compounds complexes have excellent net binding free energies.
Rights and permissions
About this article

Cite this article
Ahmad, S., Waheed, Y., Abro, A. et al. Molecular screening of glycyrrhizin-based inhibitors against ACE2 host receptor of SARS-CoV-2. J Mol Model 27, 206 (2021). https://doi.org/10.1007/s00894-021-04816-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-021-04816-y