
Abstract
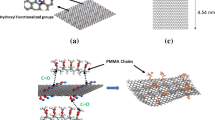
Understanding the interaction between graphene and polymers is of essential interest when designing novel nanocomposites with reinforced mechanical and electrical properties. In this computational study, the interaction of pristine graphene (PG) and graphene oxide (GO) with a series of functional groups, representative of the functionalised buildings blocks occurring in different polymers, and attached to aliphatic and aromatic chains, is analyzed using dispersion-corrected semi-empirical methods (PM6-D3H4X) and density functional theory calculations with empirical dispersion corrections. Functional groups include alkyl, hydroxyl, aldehyde, carboxyl, amino and nitro groups, and the binding energies of these groups with graphene derivatives (PG and GO) are determined. Nitro- and carbonyl groups display stronger interactions in both aliphatic and aromatic chains. The importance of dispersion-type and non-covalent interactions (NCI) in general, which typically, double the interaction energies, is revealed. The results are interpreted in an extensive NCI analysis in order to characterize the different types of NCI, providing a better understanding of the nature of the interaction (π–π stacking, CH–π bonding, H-bonding and lone pair–π interaction) at stake. In order to highlight the influence of polymer structure/conformation on top of that of their functional groups, the binding of three polymers, polyethylene (PE), polystyrene (PS) and polyvinylidene fluoride (PVDF), on pristine graphene is also investigated. Our calculations indicate that, although all polymers exhibit evident attractive interactions with the graphene sheet, the overall interaction is strongly influenced by the specific polymer structure. Thus, three main conformations of PVDF (the so-called α, β and γ, ε conformations) are analyzed and we find that, although the α-conformer with a trans-gauche-trans-gauche (TGTG’) conformation is the lowest energy conformer, the β-conformation of PVDF with the hydrogen atoms facing the graphene (“F-up”) has the strongest interaction with the graphene surface among the polymers under consideration. Taken together, our computational approach sheds light on the character and importance of non-covalent graphene-polymer functional group interactions combined with the structural/conformational properties of the polymer, which are at stake in the design of novel nanocomposites with reinforced mechanical and electrical properties.













Similar content being viewed by others


References
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666–669
Balandin AA, Ghosh S, Bao W, Calizo L, Teweldebrhan D, Miao F, Lau CN (2008) NanoLetter 8:902–907
Lee C, Wei X, Kysar JW, Hone J (2008) Science 321:385–388
Stankovich S, Dikin DA, Dommett GH, Kohlhaas KM, Zimney EJ, Stach EA, Piner RD, Nguyen ST, Ruoff RS (2006) Nature 442:282–286
Kuilla T, Bhadra S, Yao D, Kim NH, Bose S, Lee JH (2010) Prog Polym Sci 35:1350–1375
Park S, Ruoff RS (2009) Nat Nanotechnol 4:217–224
Qin W, Li X, Bian WW, Fan XJ, Qi J-Y (2010) Biomaterials 31:1007–1016
Guo Y-N, Lu X, Wenig J, Leng Y (2013) J Phys Chem C 117:5708–5717
Ganji MD (2009) Diam Relat Mater 18:662–668
Rajarajeswari M, Iyakutti K, Kawazoe Y (2011) J Mol Model 17:2773–2778
Leenaerts O, Partoens B, Peeters FM (2008) Phys Rev 77:125416
Minoia A, Chen L, Beljonne D, Lazzaroni R (2012) Polymer 53:5480–5490
Kawai H (1969) Jpn J Appl Phys 8:975
Nalwa H (1995) Ferroelectric polymers, vol 353. Dekker, New York
Hasegawa R, Takahashi Y, Chatani Y, Tadokoro H (1972) Polym J 3:600
Lovinger AJ (1981) Polymer 22:412–413
Ramasundaram S, Yoon S, Kim KJ, Park C (2008) J Polym Sci B Polym Phys 46:2173–2187
Matsushige K, Nagata K, Imada S, Takemura T (1980) Polymer 21:1391–1397
Yang D, Chen Y (1987) J Mater Sci Lett 6:599–603
Lund A, Gustafsson C, Bertilsson H, Rychwalski RW (2011) Compos Sci Technol 71:222
Pertsev NA, Zembilgotov AG (1994) Macromolecular 27:6936–6941
Shen B, Zhai W, Chen C, Lu D, Wang J, Zheng W (2011) ACS Appl Mater Interfaces 3:3103–3109
Vondrasek J, Kubai T, Jenney FE, Adams MWW, Kosizek M, Cerny J, Sklenar V, Hobza P (2007) Chem Eur J 13:9022–9027
Hobza P, Rezac J (2016) Chem Rev 116:4911–4912
Jain A, Ramanathan V, Sankararamakrishnan R (2009) Protein Sci 18:595–605
Stewart JP (2007) J Mol Model 13:1173
Johnson ER, Keinan S, Mori-Sanchez P, Chaudret R, Contraras-Garcia J, Cohen AJ, Yang W (2010) J Am Chem Soc 132:6498
Contreras-García J, Boto RA, Izquierdo-Ruiz F, Reva I, Woller T, Alonso M (2016) Theor Chem Accounts 135:242
Nart C, Maroun Z, Bota RA, Chaudret R, Bonet ML, Piquemal JP, Contreras Garcia J (2016) In: Chauvin R, Lepetit C, Silvi B, Alikhani E (eds) Applications of topological methods in molecular chemistry. Springer, Heidelberg
Alonso M, Woller T, Martín-Martínez FJ, Contreras-García J, Geerlings P, De Proft F (2014) Chem Eur J 20:4931–4941
Hajgato B, Güryel S, Blairon J, Dauphin Y, Miltner H, Van Lier G, De Proft F, Geerlings P (2012) J Phys Chem C 116:22608–22618
Güryel S, Hajgato B, Dauphin Y, Blairon JM, Miltner H, De Proft F, Geeerlings P, Van Lier G (2013) Phys Chem Chem Phys 15:659–665
Hajgato B, Güryel S, Dauphin Y, Blairon JM, Miltner H, Van Lier G, De Proft F, Geerlings P (2013) Chem Phys Lett 546:37–40
Hajgato B, Güryel S, Dauphin Y, Blairon JM, Van Lier G, Miltner HE, De Proft F, Geerlings P (2014) Indian J Chem Spec Issue Chem React Theory 53A:1058–1063
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci GA, Petersson B, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski SW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09 (revision B.01). Gaussian, Inc, Wallingford
Rezac J, Fanfrlik J, Salahub D, Hobza P (2009) J Chem Theory Comput 5:1749–1760
Rezac J, Riley KE, Hobza P (2012) J Chem Theory Comput 8:4285–4292
Korth M, Pitonak M, Rezac J, Hobza P (2010) J Chem Theory Comput 6:344–352
Hostas J, Rezac J, Hobza P (2013) Chem Phys Lett 568:161–166
Rezac J, Hobza P (2011) Chem Phys Lett 506:286–289
Stewart JJ (2012) P. MOPAC2012, Stewart computational chemistry. Colorado Springs, http://OpenMOPAC.net. Accessed 2012
Grimme S (2006) J Comput Chem 27:1787–1799
Vázquez-Mayagoitia Á, Sherrill CD, Aprà E, Sumpter BG (2010) J Chem Theory Comput 6:727–734
Peverati R, Baldridge KK (2008) J Chem Theory Comput 4:2030–2048
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Van Duyneveldt FB, Van de Rijdt JGCM, Van Lenthe JH (1994) Chem Rev 94:1873–1885
Lipinski J, Chojnacki H (1981) Int J Quantum Chem XIX:891–900
Contreras-García J, Johnson ER, Keinan S, Chaudret R, Piquemal J-P, Beratan DN, Yang W (2011) J Chem Theory Comput 7:625–632
Elkington PA, Curthoys G (1969) J Phys Chem 73:2321–2326
Zacharia R, Ulbricht H, Hertel T (2004) Phys Rev B 69:155406
Grimme S (2008) Angew Chem Int Ed 47:3430–3434
Ran J, Hobza P (2009) J Chem Theory Comput 5:1180–1185
Kuila T, Bose S, Khanra P, Mishra AK, Kim NH, Lee JH (2012) Carbon 50:914–921
Breneman CM, Wiberg KB (1990) J Comput Chem 11:361–373
Hehre WJ, Radom L, Schleyer PR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Ramanathan T, Abdala AA, Stankovich S, Dikin DA, Herrera-Alonso M, Piner RD, Adamson DH, Schniepp HC, Chen X, Ruoff RS, Nguyen ST, Aksay IA, Prud’Homme RK, Brinson LC (2008) Nat Nanotechnol 3:327–331
Kim DY, Singh NJ, Kim KS (2008) J Chem Theory Comput 4:1401–1407
Alonso M, Pinter B, Woller T, Geerlings P, De Proft F (2015) Comput Theor Chem 1053:150–164
Pinter B, Nagels N, Herrebout WA, De Proft F (2013) Chem Eur J 19:519–530
Huang X, Qi X, Boey F, Zhang H (2012) Chem Soc Rev 41:666–686
Vagione HJ, Gómez MA, Martínez G (2009) Macromolecular 42:6331–6334
Campbell K, Gurun B, Sumpter BG, Thio YS, Bucknall DG (2011) J Phys Chem B 115:8989–8995
Perdew JP, Wang Y (1992) Phys Rev B 45:13244–13249
Yu S, Zheng W, Yu W, Zhang Y, Jiang Q, Zhao Z (2009) Macromolecular 42:8870–8874
Bohlen M, Bolton K (2014) Phys Chem Chem Phys 16:12929–12939
Acknowledgements
The authors would like to acknowledge the financial support of SOLVAY, S.A. In addition, the authors acknowledge support from the COST Materials, Physical and Nanosciences (MPNS) Action MP0901: “Designing Novel Materials for Nanodevices—From Theory to Practice (NanoTP)”. M. A. thanks the Fund for Scientific Research , Flanders (FWO-12F4416N), for a postdoctoral fellowship, and the Free University of Brussels (VUB) for financial support.
The authors, and in particular P.G. and F.D.P., want to dedicate this contribution to their colleague Henri Chermette to whom this special issue is dedicated. Henri has been a very fine colleague, almost a friend, for so many years, a true companion in the search for what Density Functional Theory can really contribute to chemistry. That the recently developed and completetely density based NCI method may help chemists in the design of nanocomposites may be an illustration where the Density has led us, and will lead us.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper belongs to Topical Collection Festschrift in Honor of Henry Chermette
Appendix
Appendix

NCI analysis of H-nitro with GO and PG. The gradient isosurfaces (s = 0.5 a.u.) are colored on a BGR scale according to the sign (λ2)ρ over the range −0.02 to 0.02 a.u.
Rights and permissions
About this article

Cite this article
Güryel, S., Alonso, M., Hajgató, B. et al. A computational study on the role of noncovalent interactions in the stability of polymer/graphene nanocomposites. J Mol Model 23, 43 (2017). https://doi.org/10.1007/s00894-017-3214-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3214-2