
Abstract
Parylene C, poly(chloro-para-xylylene) is the most widely used member of the parylene family due to its excellent chemical and physical properties. In this work we analyzed the formation of the parylene C film using molecular mechanics and molecular dynamics methods. A five unit chain is necessary to create a stable hydrophobic cluster and to adhere to a covered surface. Two scenarios were deemed to take place. The obtained results are consistent with a polymer film scaling growth mechanism and contribute to the description of the dynamic growth of the parylene C polymer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parylene is the trade name for poly(para-xylylene) derivatives and has been around for more than half a century. In 1947 the deposition of thin polymeric films of poly(para-xylylene) onto a surface using a gaseous precursor was first observed by Szwarc [1]. Today it is typically deposited by the Gorham method [2] from [2,2] paracyclophane [3, 4]. There are a number of derivatives and isomers of parylene; however, only four types are commercially available: parylene N, C (see Fig. 3a), and D with none, one, and two substituted chlorine atoms per aromatic ring, respectively. The fourth type is SCS parylene HT®, the newest commercially available parylene which replaces the alpha hydrogen atom of the N dimer with fluorine [5]. Poly(chloro-para-xylylene) (parylene C) is the most widely used member of the parylene family due to its excellent chemical and physical properties. It is used in a broad range of areas including electronics, medicine, aerospace, aviation, and the automotive industry [6–8].
The steps of the chemical vapor deposition process of parylene consist of: (A) the sublimation of the dimer in a sublimation furnace, (B) the cracking of the dimer into monomer subunits in the pyrolysis furnace, (C) the transport of the monomer into the deposition chamber, (D) diffusion of the monomer, (E) adsorption of the monomer into the substrate, (F) surface migration, and (G) chemical reaction [6, 9]. While steps A, B, and C have little influence on the polymer film structure, the last four steps are crucial.
Three main ideas conducted separately are known as ways to improve the functionalities and physicochemical properties of parylene film. The first method is chemical modification of [2,2] paracyclophanes before polymerization where subsequent vapor deposition leads to functionalized chains. The second method is to use substrates which are capable of reacting with monomers or biradical chains solely during the polymerization, which leads to the modification of the layer over the reactive substrate. The former method requires the changing of process parameters, as new types of monomers influence the parylenes structure. The third method is to modify the conditions of the vapor deposition process [10–12]. Therefore, a detailed understanding of the last four steps of the chemical vapor deposition process of parylene is essential toward formulating a better design of the structure of parylene film.
To date, several growth mechanisms of the polymer film have been proposed [9, 13–16]. There are also a number of experimental [7, 11, 16–18] and theoretical [12, 19–21] studies on parylene film structure. However, most evidence argues for the scaling growth mechanism proposed by Cetinkaya et al. [11]. According to this mechanism, three steps of polymer film growth can be identified. In the first, the vapor flux arrives at an oblique angle and leads to the formation of higher surface features (columns) due to a geometric shadowing process. In the second, the approaching monomers are captured by the columns and the valleys are not filled. In this case, the film gets thicker and the columns wider. The enhanced reactivity and diffusion encourages the monomers to search for the nearest end of the polymer. Finally, the number of columns decreases as the film gets thicker until a critical thickness is reached. Bowie and Zhao [12] used a simple two-dimensional model Monte Carlo simulation of vapor deposition polymerization to show that the polymer chain length, polymer film thickness and density, and polymer structure depends on the ratio of diffusion coefficient to deposition rate. Molecular dynamics simulations were applied to study the structure and dynamics of poly(p-xylylene) in the bulky phase and of isolated chains at different temperatures using a simple empirical potential [19]. These simulations reproduced experimentally known changes in polymer density as a function of temperature and thermal expansion coefficient, and showed that the temperature disordering of the initial alpha-crystaline structure is close to the experimentally measured temperature of transition between alpha and beta polymorphs of poly(p-xylylene). Theoretical calculations performed by Sroka-Bartnicka et al. [19] shows that a change in the chain conformation of parylene N causes a change in the isotropic values of the 13C chemical shifts by a few ppm, while intermolecular interactions bring about a difference of approximately 2 ppm in 13C δiso only.
Herein, we report the results of our studies on first stages of parylene C polymerization on the water surface. The parylene polymer can be deposited on liquid surfaces such that the liquid can be used as a solid substrate. The resulting structure remains thermally and chemically stable, and the liquid is hermetically sealed under the parylene layer. The parylene layer does not exert any stress on the liquid surface and hence perfectly replicates the surface of the liquid [22–24].
Classical molecular dynamics can give insight into the mechanism of growth of the structure of the polymer film, diffusion coefficient, the nature of clustering, and the effect of adhesion. Also, conformations of the chains were tracked throughout the dynamics to find the relationship between the length, cluster stability, and the structure in the polymer film in its formation process.
Methods
Since parylene C (see Fig. 3a) is the most widely used among the parylene family, this molecule was selected for simulation. In the first step, parylene C was parameterized using the antechamber program from the AMBER v 9.0 [25, 26] package. The only available torsion angle was set as default X-CT-CT-X parameter as available in AMBER [27]. A TIP3P [28] water box (110 × 110 × 40 Å approximate size) was then built using the leap program from AMBER (v. 9.0 package). The simulation was performed on the water’s surface based on very recent information regarding the following only marginally described phenomenon: The parylene polymer can be deposited on liquid surfaces such that the liquid can be used as a solid substrate. The resulting structure remains thermally and chemically stable, and the liquid is hermetically sealed under the parylene layer. The parylene layer does not exert any stress on the liquid surface and hence perfectly replicates the surface of the liquid [22–24].
Seven different molecular systems with a polymer length of 1, 2, 3, 4, 5, 11, and 121 units were built under the assumption that one parylene unit can occupy a surface area of 10 × 10 Å2. The distance between two vicinal cells (with the aromatic ring placed in the center, as measured for the α-form of parylene N by Wunderlich et al. [29] was 10.64 Å. The same distance measured for parylene C by Lakhtakia et al. [30] was 12.69 Å (monoclinic unit cell a = 596 pm, b = 1269 pm, c = 666 pm and β = 135.2°) [30]. The calculated parylene C density (using cell size by Lakhtakia et al.) is 0.643 g cm-3 (no further symmetry information is given). Using the symmetry information from [19] the density is 1.286 while the experimentally measured density is 1.289 g cm-3 [31]. In our calculation the observed density in a cluster was about 1.286 g cm-3. The slight differences are most probably caused by the difference in the used Lennard-Jones parameters. Additionally observed differences may be caused by the fact that planar parylene C films produced conventionally are typically only about 45-60% crystalline [6, 7]. The length of the chain axis is consistent with available crystallographic data within a difference of 0.2 Å [30]. (The length of the chain axis measured by Lakhtakia et al. was 6.66 Å, whilst in our model it is 6.87 Å.) In our first simulated system, as a consequence of the assumptions taken and the water box built, 121 parylene C monomers were placed symmetrically on the 110 × 110 Å2 water surface. (According to our results (not shown) the final conformation of parylene chain does not depend on the starting orientation.) In the remaining computed systems, we attempted to maintain the aforementioned 121 units per calculation. In a second system, we employed 60 dimers, and in a third, 40 trimers, and so on, in a similar fashion. The last computed system was one entailing a single chain with a length of 121 units. This approach is justified by the fact that during parylene polymerization, the polymer chain grows one unit at a time [6, 32]. To compare the results with those containing a higher degree of the parylene concentration (and therefore deposition speed), we additionally computed the aforementioned systems with a double layer (i.e., concentration) of parylene molecules. This results in 14 computed systems.
In all cases, 2 ns molecular dynamics with a 1 fs time step, constant volume, periodic boundary conditions, and the SHAKE algorithm [33] on the hydrogen atoms were applied. For long-range electrostatic interactions, the PME (Particle Mesh Ewald) method was employed [34]. To limit the direct space sum for PME, a 12 Å cutoff was used. Non-bonding interactions were updated every 25 steps.
To maintain the flat shape of the water surface, we increased the Z-axis size of the computed box from 40 Å to 100 Å in order to gain an average density of about 0.4 g cm-3 as described in [35]. During the first 250 ps the systems were linearly heated from 10 to 300 K and further simulated in a temperature of 300 K without any constraints. (Parylene C spontaneously polymerizes on surfaces maintained below 90 °C, usually the condensation is made at room temperature [2, 6].) Data were collected every 1000 steps.
Results and discussion
The example systems after 2 ns simulations are presented on Fig. 1 (for more details see Supplementary data). As it is shown, hydrophobic oligomers are grouped into smaller or larger clusters which dimensions depending on the number of units in the chain and the concentration. What is also interesting is the fact that for longer chains, the interactions are sufficiently large to keep the chains ordered. This means that when the chains are of an adequate length, they keep together throughout their entire body length (see Fig. 1 n = 11). This in turn suggests that with sufficient parylene chain growth and an accordingly not too low density (which depends on the conditions in the polymerization chamber), the structure of the resultant parylene film can be modulated to adopt a more or less regular conformation; this effect was confirmed experimentally [12, 17]. We further speculate that with a high deposition ratio, the newly formed chains will not have enough time to adopt a compact conformation. With a high deposition ratio, the core of the newly formed column is irregular as is shown with n = 2 and 3 on Fig. 1, where chain elongation of the polymer occurs so as to merit structure roughening. With a low deposition ratio, the core of the newly formed column is regular, as is shown on Fig. 1 for n = 5 and 11 (newly formed oligomers have enough time for relaxation); here, polymer chain elongation results in compact structure.

Simulated systems after 2 ns molecular dynamics. Water surface contained about 8800 water molecules and 110x110x42 Å approximate Q3 size. The distance between water surface and the parylene molecule was set to about 5 and 15 Å in a first and second layer, respectively
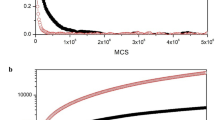
Four types of parameters were used to analyze the obtained results - the angle between the surface created by the aromatic ring of the unit and the water surface, the dihedral angle between two connected parylene units, the average distance of the parylene unit to the water surface and the mobility of the parylen on the water surface. The results are summarized in Figs. 1, 2 and 3 (for more information see Supplementary data).

The angle between the surface created by the aromatic ring of every unit and the water surface. (a) Definition of the angle. (b) Top: the average angle. Bottom: the angle distribution. The angle population was divided for three ranges 0–30 – red, 30–60 – green and 60–90 – blue and plotted as a percentage of distribution

Dihedral angle between the aromatic rings of two vicinal units. (a) Definition of the angle. (b) Top: the average angle. Bottom: the angle distribution. The angle population was divided for three ranges 0–30 – red, 30–90 – green, 90–150 – blue and 150– 180 – orange and plotted as a percentage of distribution
According to our results the average angle between the parylene unit and the water surface depends strongly on the length of the simulated oligomer. With chain elongation, the average angle increases (see Fig. 2) from 0° (at the beginning of the simulation) to a maximum of 70° (in the system with a chain length of 11 units). While there are no large differences between systems with a chain length from 1 to 4 units, we observed a rapid increase in the population of units with angles between 60 and 90° in a system with a chain length of 5 units (see Fig. 2, n = 5). This led us to postulate that the minimum number of parylene units in a chain which are necessary to adhere to a covered surface, and to create a stable hydrophobic cluster, is five. The differences in angle population for 11 and 121 unit systems is caused by the steric effect of chain bending. Some of the units placed on the chain bend to prevent the valence angle distortion in order to adopt the parallel conformation to the water surface. Our results show that the average angle between the parylene unit and the water surface also depends on the polymer concentration. This effect can be explained by the formation of the second layer of polymer. This is well visible in the following systems: n = 5, n = 5 × 2, n = 11 and n = 11 × 2. In one layer systems, there are not enough molecules to form the second layer of the polymer film. Additionally, in the n = 5x2 system, we observed a cluster of approximately 44 × 64 × 9 Å in dimension. According to Eq. 1 in ref. [11], the diameter of a 9 Å high column formed by parylene molecules should be circa 68 Å. (Characterization of the structure of parylene C revealed a growth mechanism obeying the power-law scaling. Power-law scaling proposes a columnar growth by relating its diameter to its height with an allometric equation as follows: d = chp, where d is the column diameter, c and p are constants, and h is the height of a column. For parylene C the value of constants is as follows: c = 55.20 ± 2.23 and p = 0.11 ± 0.01. [11]). With comparison to our results, this calculation is 4 Å in error. However, with a small number of simulated molecules, we find this error to be within an acceptable margin. This effect is confirmed experimentally by Lee et al. [16] (in the first step of the polymer deposition process, islands with rounded edges are formed, see Fig. 1 in ref. [16]).
The analysis of the dihedral angle between vicinal units shows that this aspect does not depend on the polymer concentration. The ratio of cis-like to trans-like conformations in systems with a chain length from 2 to 5 is almost equal, while the population of gauche-like conformations becomes smaller. In the system with a chain length of 11 units, only about 10% of the angle population become trans-like, while the rest remains in a cis-like conformation. These results can be explained by the speed of hydrophobic cluster formation. With longer chains, the speed of hydrophobic cluster formation is much higher than in the case of short chains; additionally, the formed clusters are much more stable. While the cluster is forming, the polymer chains located in the local vicinity are close enough to prevent dihedral angle rotation. In systems with long chains, we observed a slight increase in rotation parameter. This is caused by units placed on chain bends which do not participate in cluster formation. This result is in agreement with the rotation profile of the dihedral angle between aromatic rings presented by Wunderlich et al. [36], and also with the results obtained for parylene N by de Leeuw et al. (see Fig. 9a and 14 in ref. [19]). According to Sroka-Bartnicka et al. [20], the difference in the internal energy between the “straight” and “bent” conformations (calculated using the GIAO B3LYP [37] hybrid functional and the 6-311++G** basis set as implemented in the Gaussian 03 program [38]) is about 1.9 kcal mol-1, as calculated for a trimer. 1.9 kcal mol-1 is not enough to prevent free conformational change, which in turn indicates that chain interactions are essential in maintaining the “straight” conformation.
The average distance of the parylene unit to the water surface was measured as the distance of the center of mass of every parylene unit to the water surface (see Table 1). Our results reveal that with chain elongation, the average distance grows for the systems n = 1 – 3, also we observed a distance drop for: n = 4, n = 5, n = 11 and n = 121. For the n = 1 system, all monomers were placed on the water surface, creating only one layer. In this case, the average distance is about 4 Å. With an increase in concentration, some of the units migrated to the second layer and we observed a 1 Å growth in distance. Dimers and trimers form irregular structures, which are possible cores for the columns observed experimentally. For tetramers (n = 4 and n = 4 × 2 systems), we observed initial signs of order and stabilization of the structure with the average distance of the parylene unit to the water surface becoming about 1 Å lower than the distance in the corresponding trimer system. The average distance for the n = 5 system is the same as in n = 1, however, in this case, due to structure stabilization, the fluctuations are much lower. All simulated one layer systems with chain length greater than four create one stable layer of polymer film with an average distance of 4 Å to the water surface. In double concentration systems (×2), we observed the creation of a second layer of polymer film and an average distance between 5 and 6 Å.
The average mobility was measured as the difference in the location of the mass center of every parylene unit between two snapshots (1000 MD steps). Our results reveal that the average mobility of the parylene oligomers on the water surface depends strongly on the chain length. We observed the highest mobility in the case of monomers, and a rapid decrease of this parameter for dimers. The calculated mobility for all other systems (with a chain length of three and greater) is between 1 and 1.2 Å. This clearly shows that the mobility is connected to the polymerization mechanism of parylene, as is consistent with the results obtained by Senkevich et al. [39] and our previous work [40]. Similarly, to initiate polymerization, a diradical trimer is required. The activation energy barrier for the reaction of forming a dimer from two monomers is equal to about 15 kcal mol-1, while the difference in the energy between the substrate and product is about 13 kcal mol-1. Furthermore, the elongation reaction of the existing chain requires circa 3 kcal mol-1, whist the substrate-product energy difference is about 35 kcal mol-1 [40]. After the formation of the trimer, the average mobility decreases to below 1.2 Å, and we observe the start structure stabilization during the cluster formation process. While in the case of monomers and dimers, we observed a formation of hydrophobic clusters, nevertheless, due to the high molecular mobility, the newly formed clusters are structurally unstable. To form a stable cluster, a chain of at least 5 units is required. It is of notable mention that the energy barrier of the polymerization reaction is only twice higher than the barrier between the “straight” and “bent” conformations. The parylene oligomers exhibit very limited mobility, however, due to the fact that the polymerization reaction has a low energy barrier, the formation of the polymer film is possible at room temperature. The reverse situation is present at the beginning of polymerization in the stage of dimer formation. The activation energy barrier for the reaction is high (about 15 kcal mol-1), however, it is possible due to the high mobility of the monomers. Subsequently, during chain elongation, average mobility is further decreased and seems to stabilize to within a range of about 1 Å. We observed a limited effect of concentration of parylene C oligomers on average mobility.
Relying on our results, we propose the following scenario for the formation of parylene film structure: After the pyrolytic decomposition of p-cyclophanes into monomers, dimers are formed with an energy barrier of 15 kcal mol-1. The high energy barrier of this reaction is partially compensated by the high mobility of the monomers. The average mobility of dimers is much lower, and the energy barrier of the addition of the third unit to the dimer is about five times smaller. In the case of monomers and dimers, we observed a formation of hydrophobic clusters, however their structure was not stable. With the formation of trimers, the first stable clusters begin to appear. We can observe this process in (n = 3) where the gauche population is still high, albeit smaller than in the case of dimers (n = 2). Furthermore, there is no large difference in the gauche populations for chains 3 to 4 units, although we can observe a rapid decrease of the gauche population for a five unit chain. This is caused by the formation of stable clusters as described previously. From this point onward, chain elongation may be considered to occur via two scenarios. In the first, the polymer chain remains in the “straight” conformation, while in the second, the polymer chain starts to bend and to form a second layer of polymer film. This process is well related to the deposition speed of the monomers [12]. With high deposition speed, more chain bending occurs and the structure of the parylene film will be patchy. With low deposition speed, the number of chain bends will be smaller, and the structure of the parylene film will be uniform (see Fig. 2 in ref [12]).
Conclusions
It is known that with different conditions (rate, temperature, angle, etc.) for the parylene deposition process, different polymer film structures can be obtained [11, 17, 41]. However, it seems that diffusion [9] for this process [12] (connected with the structure relaxation time) is a key parameter in the design of polymer structure. To date, the atomic resolution of the parylene film structure is not available, and therefore theoretical data can be significantly useful in providing structural information. Today, even with still growing computational power, it is impossible to perform long range molecular dynamics with the full length of parylene chain (2000–4000 units) [42, 43]. It is also impossible to yet compute and observe real time growing of the parylene film with a critical thickness of about 5000 – 10,000 Å [11] and a growing rate of 50 Å min-1 in a temperature of about 30 °C [9].
According to our results, the average angle between the parylene unit and the water surface depends strongly on the length of the oligomer. With chain elongation, the average angle increases, and we observe a significant jump in the population of parylene units orthogonal conformation to the water surface, yet for those species whose chain is five and greater units long. Five unit chains were also the first group in which the average distance to the water surface was stable, with low fluctuations. This led us to postulate that the minimum number of parylene units in a chain, necessary for adherence to a covered surface and for creation the stable hydrophobic cluster, is 5. The analysis of the dihedral angle between vicinal units shows that this aspect does not depend on the polymer concentration; however it can depend on the polymer deposition ratio. Our results reveal that the average mobility of the parylene oligomers on the water surface depends strongly on the chain length. We observed the highest mobility in the case of monomers, and a rapid decrease of this parameter for dimers. The calculated mobility for all other systems (with a chain length of three and greater) is between 1 and 1.2 Å. We conclude that the mobility is connected to the polymerization mechanism of parylene. The parylene oligomers exhibit very limited mobility; however, due to the fact that the polymerization reaction has a low energy barrier, the formation of the polymer film is possible at room temperature. The reverse situation is present at the beginning of polymerization in the stage of dimer formation. The activation energy barrier for this reaction is high (about 15 kcal mol-1), however, it is possible due to the increased mobility of the monomers. Subsequently, during chain elongation, average mobility is further decreased, and seems to stabilize to within a range of about 1 Å. We observed a limited effect of parylene C oligomers concentration on the average mobility.
Using our theoretical calculations and built models, we propose a scenario of the formation of the structure of parylene film; the suggested pathway is consistent with the observed column growth mechanism by Demirel et al. [11] in the parylene deposition process. While the polymer chain is growing, the molecular mobility is decreasing. In the case of parylene C, the minimum number of units of chain to create a stable hydrophobic cluster and to adhere to a covered surface is five. Subsequently, with chain elongation, two possibilities arise. The polymer chain can either bend and start to form a second layer, or remains in a “straight” conformation. The selected pathway is related with the deposition speed of the monomers. With high deposition speed, more chain bends occur and the structure of the parylene film will be patchy. With low deposition speed, the number of chain bends will be lower, and the structure of the parylene film will be uniform as suggested by Bowie and Zhao [12].
References
Szwarc M (1947) Some remarks on the CH2[benzene]CH2 molecule. Discuss Faraday Soc 2:46–49
Gorham WF (1966) A New, General Synthetic Method for the Preparation of Linear Poly-p-xylylenes. J Polym Sci A 1:3027–3039
Reich HJ, Cram DJ (1969) Macro rings. XXXVII. Multiple electrophilic substitution reactions of [2.2]paracyclophanes and interconversions of polysubstituted derivatives. J Am Chem Soc 91:3527–3533
Lahann J, Langer R (2002) Novel Poly(p-xylylenes): thin films with tailored chemical and optical properties. Macromol 35:4380–4386
Specialty Coating Systems (SCS), 7645 Woodland Drive, Indianapolis, IA, 46278, USA. http://www.scscoatings.com/
Beach WF, Lee C, Bassett DR, Austin TM, Olson R (2004) “Xylylene Polymers”. In: Encyclopedia of Polymer Science and Technology, 3rd edn. Wiley, New York
Kahouli A, Sylvestre A, Ortega L, Pomni F, Yangui B, Maillard M, Berge B, Robert JC, Legrand J (2009) Structural and dielectric study of parylene C thin films. App Phys Lett 94:152901–152903
Nosal A, Izydorczyk A, Sobczyk-Guzenda A, Głuchowski L, Szymanowski H, Gazicki-Lipman M (2009) Parylene coatings on biological specimens. J Achiev Mat Manufact Eng 37:442–447
Fortin JB, Lu TM (2002) A Model for the Chemical Vapor Deposition of Poly(para-xylylene) (Parylene). Thin Films Chem Meter 14:1945–1949
Lahann J (2006) Vapor-based polymer coatings for potential biomedical applications. Polym Int 55:1361–1370
Cetinkaya M, Malvadkar N, Demirel MC (2008) Power-Law Scaling of Structured Poly(P-Xylylene) Films Deposited by Oblique Angle. J Pol Sci B Pol Phys 46:640–648
Bowie W, Zhao YP (2004) Monte Carlo simulation of vapor deposition polymerization. Surface Sci Lett 563:L245–L250
Biscarini F, Samori P, Greco O, Zamboni R (1997) Scaling Behavior of Anisotropic Organic Thin Films Grown in High Vacuum. Phys Rev Lett 78:2389–2392
Zhao YP, Fortin JB, Bonvallet G, Wang GC, Lu TM (2000) Kinetic roughening in polymer film growth by vapor deposition. Phys Rev Lett 85:3229–3232
Tsamouras D, Palasantzas G (2002) Temperature dependence of the growth front roughening of oligomer films. Appl Phys Lett 80:4528–4530
Lee IJ, Yun M, Lee SM, Kim JY (2008) Growth mechanisms of vapor-born polymer films. Phys Rev B 78:115427–115433
Demirel MC (2008) Emergent properties of spatially organized poly(p-xylylene) films fabricated by vapor deposition. Coll Surf A Physicochem Eng Asp 321:121–124
Cetinkaya M, Boduroglu S, Demirel MC (2007) Growth of nanostructured thin films of poly( p-xylylene) derivatives by vapor deposition. Polymer 48:4130–4134
Gotlib IY, Piotrovskaya EM, de Leeuw SW (2007) Molecular Dynamics Simulation of Poly(p-xylylene): Bulk Phase and a Single Molecule. J Phys Chem C 111:6613–6620
Sroka-Bartnicka A, Olejniczak S, Ciesielski W, Nosal A, Szymanowski H, Gazicki-Lipman M, Potrzebowski MJ (2009) Solid state NMR study and density functional theory (DFT) Calculations of structure and dynamics of poly(p-xylylenes). J Phys Chem B 113:5464–5472
Son SW, Ha M, Jeong H (2009) Anomalous scaling behavior in polymer thin film growth by vapor deposition. J Stat Mech (2009) P02031. doi:10.1088/1742-5468/2009/02/P02031
Bolognesi A, Botta C, Andicsova A, Giovanella U, Arnautov S, Charmet J, Laux E, Keppner H (2009) Chemical binding of unsaturated fluorenes to poly(2-chloroxylylene) thin films macromol. Chem Phys 210:2052–2057
Keppner H, Benkhaira M (2004) Patent: WO/2006/063955; Patent: HES-SO: EP 04 405778.4
Binh-Khiem N, Matsumoto K, Shimoyama I (2010) Tensile Film Stress of Parylene Deposited on Liquid. Langmuir 26:18771–18775
Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE III, DeBolt S, Ferguson D, Seibel G, Kollman P (1995) AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structures and energies of molecules. Comp Phys Commun 91:1–41
Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods R (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688
Weiner SJ, Kollman PA, Nguyen DT, Case DA (1986) An all atom force field for simulations of proteins and nucleic acids. J Comput Chem 7:230–252
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Iwamoto R, Wunderlich B (1973) Crystal structure of poly(p-xylylene) I. α-Form. J Polym Sci Poly Phys Ed 11:2403–2411
Demirel MC, Boduroglu S, Centinkaya M, Lakhtakia A (2007) Spatially Organized Free-Standing Poly(P-xylylene) Nanowires fabricated by vapor deposition. Langmuir 23:5861–5863
V&P Scientific, Inc. 9823 Pacific Heights Boulevard, Suite T, San Diego, CA 92121. http://www.vp-scientific.com/parylene_properties.htm
Szwarc M (1976) Poly-para-xylelene: Its chemistry and application in coating technology. Polym Eng Sci 16:473–479
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Physiol 23:327–341
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A Smooth Particle Mesh Ewald Metod. J Chem Phys 103:8577–8593
Godawat R, Jamadagni SN, Errington JR, Garde S (2008) Structure, stability, and rupture of free and supported liquid films and assemblies in molecular simulations. Ind Eng Chem Res 47:3582–3590
Miller KJ, Hollinger HB, Grebowicz J, Wunderlich B (1990) On the conformations of poly(p-xylylene) and its mesophase transitions. Macromolecules 23:3855- 3859
Ditchfield R (1972) Molecular orbital theory of magnetic shielding and magnetic susceptibility. J Chem Phys 56:5688–5691
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PM, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C.02. Gaussian Inc, Wallingford, CT
Gaynor JF, Desu SB, Senkevich JJ (1995) A Model for Chemical Vapor Copolymerization of p-Xylylenes with Vinylic Comonomers: Order of Initiation and Reactivity Ratios. Macromol 28:7343–7348
Smalara K, Gieldon A, Bobrowski M, Rybicki J, Czaplewski C (2010) Theoretical study of polymerization mechanism of p-xylylene based polymers. J Phys Chem A 114:4296–4303
Senkevich JJ, Desu SB (1999) Morphology of poly(chloro-p-xylylene) CVD thin films. Polymer 40:5751–5759
Errede LA, Gregorian RS, Hoyt JM (1960) The Chemistry of Xylylenes VI. Polymerization Para Xylylene. J Am Chem Soc 82:5218–5223
Beach WF (1978) A Model for the Vapor Deposition Polymerization of p-Xylylene. Macromolecules 11:72–76
Acknowledgments
This work was supported by the MULTIPOL project STREP n°033201 founded by the European Commission under the sixth Framework Program and the Polish Ministry of Science and Education, Grant: BW/8370-5-0449-0. This research was conducted using the resources of the Informatics Center of the Metropolitan Academic Network (IC MAN) in Gdańsk, and our own Linux based cluster at the Faculty of Chemistry, University of Gdańsk.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 20727 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gieldon, A., Czaplewski, C., Smalara, K. et al. Molecular dynamics simulations of the growth of poly(chloro-para-xylylene) films. J Mol Model 17, 2725–2733 (2011). https://doi.org/10.1007/s00894-011-1050-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-011-1050-3