
Abstract
Density functional theoretical calculations have been utilized to investigate the interaction of the amino acid arginine with the (100) surface of anatase and the reproduction of experimentally measured 49Ti NMR chemical shifts of anatase. Significant binding of arginine through electrostatic interaction and hydrogen bonds of the arginine guanidinium protons to the TiO2 surface oxygen atoms is observed, allowing attachment of proteins to titania surfaces in the construction of bio-sensitized solar cells. GIAO-B3LYP/6-31G(d) NMR calculation of a three-layer model based on the experimental structure of this TiO2 modification gives an excellent reproduction of the experimental value (-927 ppm) within +/- 7 ppm, however, the change in relative chemical shifts, EFGs and CSA suggest that the effect of the electrostatic arginine binding might be too small for experimental detection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interaction of TiO2 with the mutants of bacteriorhodopsin (bR) is important in the design of efficient biosensitized solar cells (BSSC). Application of biologically-derived, inexpensive light harvesting protein sensitizers for sensitizing titanium dioxide (TiO2) nano particles and fibers, instead of the traditional, toxic and expensive chemical based dyes in dye-sensitized solar cells (DSSCs), is an attractive proposition.
Protein-sensitized solar cells emulate the design of DSSCs except that the dye molecules are replaced by light harvesting biomolecules as sensitizer. The impetus for this replacement comes from potentially high efficiency of conversion that light harvesting protein complexes promise, their ability to self-assemble on a surface [1], their low cost of production by biotechnology and room temperature processing and their long lifetimes even at higher temperature (100°C) [2]. An interesting candidate for a protein sensitizer is bacteriorhodopsin (bR) which is a trans-membrane protein found in the archae Halobacterium salinarum [3]. Absorption of photon in bR occurs principally through the chromophoric group and the charge separation that occurs eventually leads to shedding of electrons. Upon absorption of light, there is a shift of electron density from the β-ionone ring of the chromophore toward the Schiff base, which is comparable to a 2.6 Å displacement of a single electron down the polyene chain [4] and is in the opposite direction of the initial photovoltage spike that is observed in bR [5]. This short duration charge shift is a critical component of the primary event because it sets in motion events that lead to the photoisomerization of the chromophore [6].
TiO2 has been in the focus of numerous investigations since the demonstration of the original DSSC photovoltaic cell [7]. Researchers have proposed the possibility of using hybrid architecture of nanoparticles and nanofibres [8–12] to facilitate better electron transport. Given the advancement in electrospinning for generating large surface-to-volume ratio nanofibres in larger-scale [13], the electrospun TiO2 nanofibres have been shown to increase solar energy efficiency up to 6.2 % in DSSC [14].
The basic hypothesis for the design of such a BSSC, for which we propose the name Renu-Seeram cell is that bR is attached by chemisorption to TiO2, and upon photoexcitation ejects electrons into the conduction band of semiconducting metal oxide completing the circuit and electrons flow from counter electrode to the conductive ITO glass plate. An iodide electrolyte recharges bR.
In order to shed some light on this interaction, we set out to investigate computationally the binding of arginine, which has been identified as a potential terminal amino acid in bR, on the (100) (or identical by symmetry the (010)) surface of TiO2 anatase [15] in order to derive a model for the interaction of TiO2 to bR mutants.
Methods
All calculations were carried out using the Gaussian 03 [16] suite of programs. Density functional theory (DFT) with the hybrid B3LYP functional [17–19] in combination with Pople’s 6-31G(d) [20, 21] was used for the optimizations of the ground state geometries of a TiO2 anatase model, arginine and a complex of arginine on anatase and the simulation of the chemical shifts. The default convergence criteria and integration grid of the program were used. The nature of all found local minimum structures was confirmed by the lack of an imaginary harmonic vibrational frequency [22]. Absolute magnetic shieldings were determined with the GIAO (gauge-including atomic orbital) method [23–25] as implemented in Gaussian 03. The obtained shielding tensors were referenced against titanium tetrachloride to yield relative chemical shifts. This approach has been used successfully for the determination of chemical 49Ti shifts previously [26]. The simulations of NMR tensor parameters were performed using the SIMPSON program [27]
Results and discussion
When a protein such as bR or its mutants is brought onto a surface such as anatase or ZnO, it will be either covalently attached or chemisorbed. It is therefore reasonable to expect subtle electronic perturbations of 49Ti chemical shift anisotropy (CSA or Δσ) and the quadrupole coupling, Cq, which provide useful tools to probe subtle interactions at the atomic level. It should be possible to compare the DFT derived changes in electric field gradient and CSA with experimental values [28].
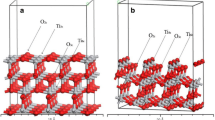
TiO2 surfaces have been thoroughly studied, both experimentally and theoretically [29–40]. Also, the adsorption of small molecules on TiO2 surface has been reported in the literature recently [41–52]. Based on previous experience with the calculation of 49Ti NMR chemical shifts [25], we have started to explore a suitable model for the TiO2 (100) surface on which the bR is attached [15]. This surface is one of the two major cleavage planes, together with the (101) surface. It possesses small grooves formed by oxygen atoms on either side of the groove along with both Ti and O atoms at the bottom. Several repeat unit cells are required for reliable and consistent data, since there are relative chemical shifts in the range of several hundred ppm for individual Ti atoms. The model employed herein to represent the (100) surface of anatase consists of 30 TiO2 units, terminated with 12 hydrogen atoms (Fig. 1). It has been taken from the crystal structure of TiO2 anatase [53] and the positions of all surface and added hydrogen atoms are kept fixed during the optimization, only allowing the amino acid to move freely on the surface.

Model of (100) TiO2 anatase surface, top (left) and side (right) views; oxygen atoms in red, titanium atoms in gray
The well-established B3LYP/6-31G(d) level of theory has been chosen because of its previously demonstrated superior performance in the 49Ti NMR determination [26]. Herein, relative chemical shifts of the Ti atoms of this model are computed compared against the available experimental NMR data. In our model there are four titanium atoms which have a “complete” oxygen-saturated environment – in the middle layer of the side-on view of Fig. 1 – which can serve as representatives of the bulk titanium atoms. These Ti atoms are calculated to have isotropic chemical shifts (relative to TiCl4) between -921 and -922 ppm, in excellent agreement with the experimental value for anatase of -927 ppm [26]. Shifts of those titanium atoms on the surface which have five oxygen contacts are in the range of -450 to -530 ppm.
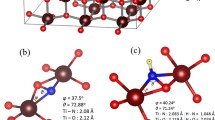
In a previous study, the positively charged arginine and lysine amino acid residues have been identified as potential binding groups as they reside at the cytoplasmic surface of bR [15]. For this reason we have chosen arginine as a model for bR in this binding investigation. Different orientations of arginine on the surface have been modeled with the most stable ones reported herein (Fig. 2): In structure 1, arginine possesses two hydrogen bonds bridging the groove, originating from each of the terminal nitrogen atoms and pointing toward the central oxygen atoms in our model. This orientation is stabilized relative to the separated TiO2 surface and arginine by about 160 kJ mol−1. The largest part of this energy can be attributed to attractive electrostatic interaction between the positively charged arginine N terminus and the surface oxygen atoms. The importance can be seen in the increasing tilting of arginine toward the surface in the following structures 2-5, thus bringing the third partially charged nitrogen atom of the guanidinium group closer to the negatively charged oxygen atoms. Structure 2 exhibits this slightly tilted position across the groove and has a third hydrogen bond which lowers the energy by an additional 60 kJ mol−1. Orientations 3 and 4 are quite similar to each other, being stabilized by 250 and 254 kJ mol−1, respectively. Both trade the docking across the gap formed by the surface oxygen atoms in favor of one intramolecular and two hydrogen bonds to oxygen atoms on the same side of the groove.

Different orientations of arginine on the anatase (100) surface model: 1 (top left), 2 (top right), 3 (middle left), 4 (middle right), 5 (bottom left) and 6 (bottom right). Hydrogen bonds are shown
The most stable arrangements are the last two presented in Fig. 2. They are 266 and 272 kJ mol−1 respectively more stable than the separate molecules and adopt an orientation which allows additional weak hydrogen contacts across the groove. Structure 5 forms such a bond at about 2.6 Å from the previously uninvolved third nitrogen atom, in addition to the two strong bonds at about 1.9 Å, while the most stable structure 6 possesses four contacts at 1.7, 2.1, 2.4 and 2.4 Å (see also Fig. 3). Both orientations retain the intramolecular hydrogen bond. These data illustrate that the attachment at an oxygen-based channel is in fact the most likely position but it appears preferentially to form as many hydrogen bonding interactions with oxygen atoms on both sides of this trough as possible, even at the expense of their individual strength. In addition, the latter structures are closer to the surface, leading to increased electrostatic stabilization.

Numbering scheme for arginine used in Table 1. The groove oxygen atoms are also shown, allowing easier identification of the hydrogen bonds
Starting from this final orientation 6, we also derived the chemical shifts for both the loaded anatase (49Ti) and the amino acid (13C). The 13C chemical shifts of arginine under the influence of the anatase attachment are given in Table 1. One can clearly see that the carbon atoms which are close to a new hydrogen bond (atoms 3 and 8) are shifted to a higher field by up to 12 ppm.
The relative 49Ti chemical shifts of the four central Ti atoms with oxygen saturation (see above) are influenced by the arginine attachment by only a few ppm (between 1 and 9 ppm, values range from -913 to -928 ppm). This appears too small to be easily detectable in a standard NMR experiment. However, the surface titanium atoms are much more strongly influenced: the induced changes in relative chemical shifts range from -83 to +93 ppm.
For comparison we have simulated the experimental parameters previously obtained for bulk anatase with those predicted by quantum mechanical calculations using the most stable orientation 6 (Fig. 2). These data are depicted in Fig. 4 for each NMR active isotope of titanium. The experimental values [26] of quadrupole coupling constant, Cq, previously measured for anatase are 4.94 and 4.04 MHz for the 47 and 49 isotopes respectively. The asymmetry parameter for each was 0.06, as these systems are nearly axially symmetric one would expect it near zero. The chemical shielding anisotropy (CSA) was 117 ppm with an asymmetry parameter of 0.1 and all three Euler angles fixed at 0° (coincident principle axis systems). These parameters are contrasted with those for the model systems in Table 2. We define the shielding anisotropy as Δσ = δ33 – ½ (δ11+δ22), where the elements of the shielding tensor are ordered as |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso|; and δiso is the trace of the tensor δ.

Ideal pulse simulations at 21.1 T (900 MHz for 1H) of solid-state NMR spectra for the experimental values for anatase (a + d), the model with no arginine on the surface (b + e), and the surface Ti atom with arginine bound (c + f) with the first column showing 49Ti and the second 47Ti NMR spectra
It is clear from the data in Fig. 4 that the level of theory seems to overestimate both the electric field gradient (EFG) and the CSA. The 47Ti (nuclear spin 5/2) data is dominated by the quadrupolar coupling which is directly proportional to the EFG tensor and therefore shows the effects of this parameter, while in the 49Ti (nuclear spin 7/2) data it is evident that the CSA interaction dominates this lineshape at the simulated magnetic field. The key point is that the effects are similar for the models with and without the arginine present. The implication is that the electrostatics of the Ti atoms on the surface are not strongly impacted by the binding of the amino acid. Although it is possible with some effort to experimentally distinguish surface atoms from the bulk, measuring such a surface effect with NMR seems difficult, despite the above mentioned calculated changes in relative chemical shifts.
Conclusions
The nature of the attachment of arginine on the anatase (100) surface is clearly based on electrostatic interaction together with hydrogen bonds. It appears that an increased number of these weak interactions lead to stabilization of the complex between our (100) anatase model and the amino acid, indicating that the attachment of bR onto a TiO2 surface is possible in the Renu-Seeram Cell [54]. Additionally, the small energetic changes between some of the found orientations suggest that one can expect dynamic behavior of arginine on the surface. The correct DFT reproduction of 49Ti NMR chemical shifts for anatase gives confidence in the reliability of the employed approach for the evaluation of this property. Detailed computational investigation of both 13C (arginine) and 49Ti (TiO2) NMR data show very little change in predicted relative chemical shifts upon binding. This is further confirmed by the simulation of the titanium spectra based on experimental and calculated data. It suggests that NMR investigation of the protein binding on titania nanoparticles seems unfeasible. Other techniques such as SERS on TiO2 [55] might give additional insight into the attachment mode.
References
Das R, Kiley PJ, Segal M, Norville J, Yu AA, Wang LS, Trammell A, Evan Reddick L, Kumar R, Stellacci F, Lebedev N, Schnur J, Bruce BD, Zhang S, Baldo M (2004) Nano Lett 4:1079–1083
Kiley P, Zhao X, Vaughn M, Baldo MA, Bruce BD, Zhang S (2005) PLoS Biol 3:e230
Oesterhelt D, Stoeckenius W (1971) Nature New Biol 233:149–152
Birge RR, Gillespie NB, Izaguirre EW, Kusnetzow A, Lawrence AF, Singh D, Song QW, Schmidt E, Stuart JA, Seetharaman S, Wise KJ (1999) J Phys Chem B 103:10746–10766
Trissl HW (1985) Biochim Biophys Acta 806:124–135
Zadok U, Khatchatouriants A, Lewis A, Ottolenghi M, Sheves M (2002) J Am Chem Soc 124:11844–11845
Regan BO, Grätzel M (1991) Nature 353:737–740
Onozuka K, Ding B, Tsuge Y, Naka T, Yamazaki M, Sugi S, Ohno S, Yoshikawa M, Shiratori S (2006) Nanotechnology 17:1026–1031
Pavasupree S, Ngamsinlapasathian S, Nakajima M, Suzuki Y, Yoshikawa S (2006) J Photochem Photobiol A: Chem 184:163–169
Pavasupree S, Ngamsinlapasathian S, Suzuki Y, Yoshikawa S (2006) J Nanosci Nanotechnol 6:3685–3692
Suzuki Y, Ngamsinlapasathian S, Yoshida R, Yoshikawa S (2006) Cent Eur J Chem 4:476–488
Asagoe K, Ngamsinlapasathian S, Suzuki Y, Yoshikawa S (2007) Cent Eur J Chem 5:1–15
Ramakrishna S, Fujihara K, Teo WE, Yong T, Ma Z, Ramaseshan R (2006) Mater Today 9:40–50
Song MY, Ahn YR, Jo SM, Kim DY (2005) Appl Phys Lett 87:113113-1-113113-2
Thavasi V, Lazarova T, Filipek S, Kolinski M, Querol E, Kumar A, Ramakrishna S, Padrós E, Renugopalakrishnan V (2009) J Nanosci Nanotech 9:1679–1687
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision C02. Gaussian Inc, Wallingford, CT
Becke AD (1988) Phys Rev A 38:3098–3100
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Hariharan PC, Pople JA (1973) Theor Chim Acta 28:213–222
Pietro WJ, Francl MM, Hehre WJ, DeFrees DJ, Pople JA, Binkley JS (1982) J Am Chem Soc 104:5039–5048
The artificially constrained hydrogen atoms of the anatase model produce distinct vibrations with negative wavenumbers; these frequencies were neglected as they are not important for the binding of arginine on the surface.
Ditchfield R (1974) Mol Phys 27:789–807
Wolinski K, Hinton JF, Pulay P (1990) J Am Chem Soc 112:8251–8260
Cheeseman JR, Frisch MJ, Devlin FJ, Stephens PJ (2000) J Phys Chem A 104:1039–1046
Koch R, Bruhn T (2006) J Mol Model 12:723–729
Bak M, Rasmussen JT, Nielsen NC (2000) J Magn Reson 147:296–330
Larsen FH, Farnan I, Lipton AS (2006) J Magn Res 178:228–236
Onda K, Li B, Zhao J, Jordan KD, Yang J, Petek H (2005) Science 308:1154–1158
Fernandez-Garcia M, Belver C, Hanson JC, Wang X, Rodriguez JA (2007) J Am Chem Soc 129:13604–13612
Asahi R, Taga Y, Mannstadt W, Freeman A (2000) J Phys Rev B: Condens Matter 61:7459–7465
Vittadini A, Casarin M, Selloni A (2007) Theor Chem Acc 117:663–671
Vittadini A, Selloni A, Rotzinger FP, Graetzel M (1998) Phys Rev Lett 81:2954–2957
Diebold U (2003) Appl Phys A 76:681–687
Hadjiivanov KI, Klissurski DG (1996) Chem Soc Rev 25:61–69
Ramamoorthy M, Vanderbilt D (1994) Phys Rev B: Condens Matter 49:16721–16727
Lazzeri M, Vittadini A, Selloni A (2001) Phys Rev B: Condens Matter 63:155409/1-155409/9
Kubo T, Orita H, Nozoye H (2007) J Am Chem Soc 129:10474–10478
Koitaya T, Nakamura H, Yamashita K (2009) J Phys Chem C 113:7236–7245
Morgan BJ, Watson GW (2009) J Phys Chem C 113:7322–7328
Topoglidis E, Cass AEG, Gilardi G, Sadeghi S, Beaumont N, Durrant JR (1998) Anal Chem 70:5111–5113
Duncan WR, Prezhdo OV (2008) J Am Chem Soc 130:9756–9762
Li J, Wu L, Zhang Y (2001) Chem Phys Lett 342:249–258
Sheka EF, Nikitina EA, Zayets VA, Ginzburg IY, Schoonman J (2005) J Nanopart Res 7:171–186
De Angelis F, Fantacci S, Selloni A, Nazeeruddin MK, Graetzel M (2007) J Am Chem Soc 129:14156–14157
Duncan WR, Prezhdo OV (2007) Annu Rev Phys Chem 58:143–184
Duncan WR, Craig CF, Prezhdo OV (2007) J Am Chem Soc 129:8528–8543
Lundqvist MJ, Nilsing M, Lunell S, Aakermark B, Persson P (2006) J Phys Chem B 110:20513–20525
Raghunath P, Lin MC (2009) J Phys Chem C 113:8394–8406
Zhang J, Zhang M, Han Y, Li W, Meng X, Zong B (2008) J Phys Chem C 112:19506–19515
Oviedo J, Sanchez-de-Armas R, San Miguel MA, Sanz JF (2008) J Phys Chem C 112:17737–17740
Morandeira A, Lopez-Duarte I, Martinez-Diaz MV, O’Regan B, Shuttle C, Haji-Zainulabidin NA, Torres T, Palomares E, Durrant JR (2007) J Am Chem Soc 129:9250–9251
Wagemaker M, Gordon JK, Van Well AA, Mutka H, Mulder FM (2003) J Am Chem Soc 125:840–848
Kang et al. have very recently reported that hydroxylation of a rutile TiO2 (110) surface significantly increases binding energies of proteins. For details see Kang Y, Li X, Tu Y, Wang Q, Ågren H (2010) J Phys Chem C 114:14496-14502
Musumeci A, Gosztola D, Schiller T, Dimitrijevic NM, Mujica V, Martin D, Rajh T (2009) J Am Chem Soc 131:6040–6041
Acknowledgments
RK gratefully acknowledges generous allocation of computer time at the Center for Scientific Computing, Uni Oldenburg and financial support by the Deutsche Forschungsgemeinschaft DFG.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Prof. Paul von Rague Schleyer on the occasion of his 80th birthday
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Supporting material available Cartesian coordinates of the B3LYP/6-31G(d)-optimized structures of anatase-arginine complexes are available in the Supporting Information (DOC 113 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Koch, R., Lipton, A.S., Filipek, S. et al. Arginine interactions with anatase TiO2 (100) surface and the perturbation of 49Ti NMR chemical shifts – a DFT investigation: relevance to Renu-Seeram bio solar cell. J Mol Model 17, 1467–1472 (2011). https://doi.org/10.1007/s00894-010-0853-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-010-0853-y