
Abstract
Density functional theory (DFT) calculations at the B3LYP/6–31G(d) level were carried out for 47 vinyl monomers with structures C1H2 = C2HR3, and the calculated quantum chemical descriptors were used to construct quantitative structure–property relationship (QSPR) models of the reactivity parameters of monomers Q and e. Stepwise multiple linear regression analysis (MLRA) and artificial neural networks (ANN) were adopted to generate the models. Simulated with the final optimum back-propagation (BP) neural networks, the results show that predicted lnQ and e values are in good agreement with experimental data, with test sets possessing correlation coefficients of 0.982 for lnQ and 0.943 for e. The proposed ANN models have better prediction ability than existing models.

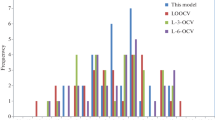
Plot of calculated versus experimental e values


Similar content being viewed by others


References
Alfrey T, Price CC (1947) J Polym Sci 2:101–109
Laurier GC, O’Driscoll KF, Reilly PM (1985) J Polym Sci: Polym Symp 72:17–26
Rogers SC, Mackrodt WC, Davis TP (1994) Polymer 35:1258–1267
Zhan CG, Dixon DA (2002) J Phys Chem A 106:10311–10325
Pan ZR (2002) Polymer Chemistry, 3rd edn. Chemical Industry, Beijing, pp 95–96
Semchikov YuD (1990) Vysokomolek Soedin A 32:243–252
Greenly RL (1980) J Macromol Sci Chem 4:427–443
Toropov AA, Kudyshkin VO, Voropaeva NL, Ruban IN, Rashidova SSh (2004) J Struct Chem 45:945–950
Karelson M, Lobanov VS, Katritzky AR (1996) Chem Rev 96:1027–1043
Yu XL, Xie ZM, Yi B, Wang XY, Liu F (2007) Eur Polym J 43:818–823
Yu XL, Yi B, Wang XY (2007) J Comput Chem 28:2336–2341
Brandrup J, Immergut EH, Grulke EA (1999) Polymer handbook, 4th edn. Wiley, New York
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, Oxford
Hobza P, Sÿponer J (1999) Chem Rev 99:3247–3276
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, MonTgomery JA Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Andres JL, Head-Gordon M, Replogle ES, Pople JA (2003) Gaussian 03, Revision B.05. Gaussian, Pittsburgh, PA
Becke AD (1993) J Chem Phys 98:5648–5652
Hehre WJ, Radom L, Schleyer PvR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Cioslowski J (1989) J Am Chem Soc 111:8333–8336
Fukui K (1975) Theory of orientation and stereoselection. Springer, New York, pp 34–39
Franke R (1984) Theoretical drug design methods. Elsevier, Amsterdam, pp 115–123
Zhou Z, Parr RG (1990) J Am Chem Soc 112:5720–5729
Mattioni BE, Jurs PC (2002) J Chem Inf Comput Sci 42:232–240
Gao JW, Wang XY, Li XB, Yu XL, Wang HL (2006) J Mol Model 12:513–520
Gao JW, Wang XY, Yu XL, Li XB, Wang HL (2006) J Mol Model 12:521–527
Lewis DFV, Ioannides C, Parke DV (1994) Xenobiotica 24:401–408
Acknowledgments
The project was supported by the Scientific Research Fund of Hunan Provincial Education Department (NO. 07C205), and the Scientific Research Fund of Hunan Institute of Engineering (NO. 0761).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, X., Liu, W., Liu, F. et al. DFT-based theoretical QSPR models of Q-e parameters for the prediction of reactivity in free-radical copolymerizations. J Mol Model 14, 1065–1070 (2008). https://doi.org/10.1007/s00894-008-0339-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-008-0339-3