
Abstract
The Acidianus hospitalis W1 genome consists of a minimally sized chromosome of about 2.13 Mb and a conjugative plasmid pAH1 and it is a host for the model filamentous lipothrixvirus AFV1. The chromosome carries three putative replication origins in conserved genomic regions and two large regions where non-essential genes are clustered. Within these variable regions, a few orphan orfB and other elements of the IS200/607/605 family are concentrated with a novel class of MITE-like repeat elements. There are also 26 highly diverse vapBC antitoxin–toxin gene pairs proposed to facilitate maintenance of local chromosomal regions and to minimise the impact of environmental stress. Complex and partially defective CRISPR/Cas/Cmr immune systems are present and interspersed with five vapBC gene pairs. Remnants of integrated viral genomes and plasmids are located at five intron-less tRNA genes and several non-coding RNA genes are predicted that are conserved in other Sulfolobus genomes. The putative metabolic pathways for sulphur metabolism show some significant differences from those proposed for other Acidianus and Sulfolobus species. The small and relatively stable genome of A. hospitalis W1 renders it a promising candidate for developing the first Acidianus genetic systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Acidianus genus consists of acidothermophiles which grow optimally and slowly in the temperature range 65–95°C and at pH 2–4 and belongs to the order Sulfolobales. Acidianus species are chemolithoautotrophic and facultatively anaerobic and are generally versatile physiologically. Depending on the culturing conditions, they can either reduce S° to H2S, catalysed by a sulphur reductase and hydrogenase, or oxidise S° to H2SO4 utilising the sulphur oxygenase-reductase holoenzyme (Kletzin 1992, 2007). In contrast to several Sulfolobus species, the genomic properties of an Acidianus species have not been analysed. The Sulfolobales have been a rich source of genetic elements, including novel conjugative plasmids (Prangishvili et al. 1998; Greve et al. 2004) and several exceptional and diverse viruses many of which have now been classified into eight new viral families (Rachel et al. 2002; Prangishvili et al. 2006; Lawrence et al. 2009).
Acidianus hospitalis W1 is the first Acidianus strain to be isolated carrying a conjugative plasmid pAH1 which is a member of the plasmid family predicted to generate an archaea-specific conjugative apparatus (Greve et al. 2004; Basta et al. 2009). These plasmids are also integrative elements and in an encaptured state have been implicated in facilitating chromosomal DNA conjugation for some Sulfolobus species (Chen et al. 2005b). A. hospitalis is also a viable host for the model Acidianus alpha lipothrixvirus AFV1, a filamentous virus carrying exceptional claw-like structures at its termini which is currently the subject of detailed structural studies (Bettstetter et al. 2003; Goulet et al. 2009). Infection of A. hospitalis with AFV1 was shown to lead to a loss of the plasmid pAH1 and this contrasts with observations in bacteria where endogenous plasmids tend to determine the fate of an incoming phage (Basta et al. 2009).
In order to study further the metabolic capability of an Acidianus species and to examine the molecular mechanisms involved in virus–plasmid–host interactions, it was important to sequence and annotate the A. hospitalis genome. To date, most genomic studies of the Sulfolobales have concentrated on Sulfolobus species that have revealed relatively large genomes generally exhibiting high levels of transposable and integrated genetic elements, as well as considerable genetic diversity (Guo et al. 2011). Analysis of the A. hospitalis genome revealed a minimally sized chromosome that appeared relatively stable with few transposable elements and no evidence of recent integration events, apart from the reversible integration of pAH1 into a tRNAArg gene (Basta et al. 2009). Potentially, therefore, A. hospitalis W1 could provide a suitable host for developing genetic systems for the Acidianus genus.
Materials and methods
Genome sequencing and gap closure
Genomic DNA of A. hospitalis was sequenced using a Roche 454 Genome Sequencer FLX instrument (Titanium) with an average 19-fold coverage. All useful reads were initially assembled into seven contigs (>500 bp) using the Newbler assembler software (http://www.454.com/). Gaps were closed by a Multiplex PCR strategy and PCR products were gel purified and sequenced using an ABI3730 DNA sequenator. Raw sequence data were assembled into contigs using phred/phrap/consed software and the final consensus quality for each base was above 30 (http://www.phrap.org).
Sequence analysis and gene annotation
Initially, ORFs were predicted using the programmes Glimmer and FgeneSB and protein function predictions were obtained from the following searches: (1) homology searches in the GenBank (http://www.ncbi.nlm.nih.gov/) and UniProt protein (http://www.ebi.ac.uk/uniprot/) databases, (2) function assignment searches in the Sulfolobus database (http://www.Sulfolobus.org/), and (3) domain or motif searches in the local CDD database (http://www.ncbi.nlm.nih.gov/cdd/), the InterPro and the Pfam databases. The KEGG database (http://www.genome.jp/kegg/) was used to reconstruct metabolic pathways in silico. Membrane proteins were predicted by Phobius, TMHMM and ConPred II programmes. Secretory proteins were divided into two groups; those with a signal peptide were predicted using the SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP/) and non-classical secretory proteins, lacking a signal peptide, were predicted by the SecretomeP 2.0 programme (http://www.cbS.dtu.dk/services/SecretomeP/). Transporters were predicted by searching the TCDB database (http://www.tcdp.org) using BLASTP with E values lower than 1e-05. Insertion sequence (IS) elements and transposases were identified by BLASTN searches against the IS Finder database (http://www-is.biotoul.fr/). The MITE-like elements were detected using the programme LUNA (Brügger K, unpublished). Potential frameshifts were checked by sequencing after manual annotation and any remaining frameshifts were considered to be authentic. tRNA genes and their introns were identified using tRNAScan-SE (Lowe and Eddy 1997). All annotations were manually curated using Artemis software (Rutherford et al. 2000). Start codons for single genes and first genes of Sulfolobus operons were generally located 25–30 bp downstream from the archaeal hexameric TATA-like box. Only genes within operons were preceded by Shine–Dalgarno motifs, where GGUG dominated (Torarinsson et al. 2005). Where alternative start codons occur, a selection was made on the basis of experimental data when available or on its location relative to a putative promoter and/or Shine–Dalgarno motif. The genome sequence accession number at Genbank/EMBL is CP002535.
Results
Genomic properties
The A. hospitalis genome consists of a circular chromosome of 2,137,654 bp and a circular conjugative plasmid pAH1 of 28,644 bp. The chromosome has a GC content of 34.2% and carries 2,389 predicted open reading frames (ORFs), of which about half are assigned putative functions with many of the conserved hypothetical proteins being archaea-specific or specific to the Sulfolobales. About 320 of the encoded proteins are putative membrane proteins and a further 182 are predicted to be secretory proteins. The plasmid sequence is identical to that of the conjugative plasmid pAH1 isolated earlier from the A. hospitalis strain W1, except that it is 4 bp shorter (Basta et al. 2009).
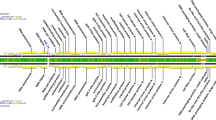
Comparison of the A. hospitalis genome with those of other members of the Sulfolobales provided no evidence of extensive conservation of gene synteny, in contrast to that observed for large regions of several Sulfolobus genomes (Guo et al. 2011), and consistent with A. hospitalis being relatively distant phylogenetically from these strains (Basta et al. 2009). Nevertheless, the genome carries two major regions that are predicted to be relatively labile. They extend approximately from positions 75,000–444,500 and from 1,300,000–1,870,000 and carry most of the transposable elements, all of the CRISPR loci and cas and cmr family genes, most of the vapBC toxin–antitoxin gene pairs, and many genes involved in transport-related functions and metabolism, as well as a degenerate fuselloviral genome (Fig. 1). These two regions lack genes essential for informational processes including DNA replication, transcription and translation and they appear to constitute sites where non-essential genes are collected, interchanged, exchanged intercellularly and where genetic innovation may occur, similarly to a single variable region observed in several Sulfolobus genomes (Guo et al. 2011).

The Y component of a Z curve plot for the A. hospitalis chromosome showing the three putative replication origins. The positions of the cdc6-3 gene (origin 2), cdc6-1 gene (origin 3) and the whiP/cdt1 gene (origin 1) are indicated as well as locations of the ribosomal RNA genes, the CRISPR-based systems, transposable elements of the IS200/605/607 family, and vapBC antitoxin–toxin gene pairs
Three origins of chromosomal replication, demonstrated experimentally for Sulfolobus species (Robinson et al. 2004; Lundgren et al. 2004), were also predicted to occur in the Acidianus genome. The Y component of a Z curve analysis (Zhang and Zhang 2003) revealed two major peaks corresponding to the cdc6-3 gene (Ahos0001), and the whiP/cdt1 gene (Ahos1370) and a broader peak coinciding with the cdc6-1 gene (Ahos0780) (Fig. 1), where the three genes encode putative replication initiators (Robinson and Bell 2007). The sequences of the cdc6 genes and whiP gene are quite conserved relative to the S. solfataricus and S. islandicus genomes, as is the synteny of the flanking genes except for the region immediately downstream from cdc6-3.
Integrated genetic elements
Integration of genetic elements, generally fuselloviruses or conjugative plasmids at tRNA genes, occurs commonly for genomes of the Sulfolobales (She et al. 1998; Guo et al. 2011). Most integration events occur via a reversible archaea-specific mechanism whereby the integrase gene partitions into two sections which border the integrated element and the N-terminal-encoding region carrying the intN sequence overlaps with the tRNA gene (Muskhelishvili et al. 1993). Elements that become encaptured within the chromosome subsequently degenerate and are gradually lost, but will nevertheless leave a trace because the intN fragment overlapping the tRNA gene is generally retained (She et al. 1998) (Table 1).
Earlier plasmid pAH1 was sequenced and shown to integrate reversibly into a tRNAArg gene (Basta et al. 2009). Genome sequencing of A. hospitalis revealed that a low fraction of reads matched to the junctions of the integrated plasmid whilst the majority matched the unpartitioned integrase gene of pAH1, consistent with both integrated and free forms being present in the culture. The integration site of pAH1 was located at genome positions 1,075,876–1,075,946 bp within the gene of tRNAArg [TCG] (Table 1). In addition, the chromosome carries remnants of integrated elements adjoining another five intron-less tRNA genes, each consisting of a few genes or pseudogenes (Table 1). Three derive from fuselloviruses, one from a pDL10-like plasmid of the pRN family of cryptic plasmids (Kletzin et al. 1999) and another originates from an unknown element (Table 1). Whether these all derive from single integration events remains unclear because, in principle, successive integrations can occur at a given tRNA gene (Redder et al. 2009). An additional 8–10 genes and pseudogenes, most of which are fusellovirus-related, are clustered distantly from a tRNA gene and they may have become displaced from one of the three tRNA-integrated elements.
Transposable elements
The A. hospitalis genome carries five IS elements belonging to the IS200/607 family, only three of which carry intact transposase genes, and there are 11 copies of orphan orfB elements of the IS605 family, 10 of which carry intact orfB genes. None of these elements carry inverted terminal repeats and they all appear to be transposed by “cut-and-paste” mechanisms, with the orfB elements, at least, transposing via circular single stranded intermediates and inserting after TTAC sequences (Filée et al. 2007; Ton-Hoang et al. 2010).
Sulfolobus genomes generally carry IS elements from a wide variety of families most of which carry inverted terminal repeats and are mobilised by “copy-and-paste” mechanisms, and tend to be lost by gradual degeneration and not by deletion (Blount and Grogan 2005; Redder and Garrett 2006). None of these IS element classes were detected in the A. hospitalis genome and this suggests that the genome has rarely, if ever, taken up any of these IS element classes.
A new class of MITE-like elements
Although none of the MITE elements that are common to other Sulfolobus genomes were detected (Redder et al. 2001; Guo et al. 2011), the A. hospitalis genome carries 10 copies of a repeat sequence resembling a MITE-like element (Fig. 3). At one end, it carries a short open reading frame corresponding in amino acid sequence to the downstream end of an OrfB protein (Fig. 3). The conserved terminal sequence and the internal similarity to the orfB element suggests that it could be a transposable element. This supposition is reinforced by the presence of 10 full copies in the genome (and a few degenerate copies), and also by the presence of multiple copies in some Sulfolobus and other crenarchaeal genomes (unpublished data).
Non-coding RNAs
Many untranslated RNAs have been characterised experimentally for different Sulfolobus species using a variety of techniques including probing cellular RNA extracts for K-turn-binding motifs and generating cDNA libraries of total cellular RNA extracts, as well as numerous antisense RNAs (Tang et al. 2005; Omer et al. 2006; Wurtzel et al. 2010). Most of these RNAs were characterised for partial sequence and nucleotide length, and several were detected by more than one experimental approach. Based on the genome sequence comparisons and gene contexts, 23 putative conserved non-coding RNAs were annotated in the A. hospitalis genome. Genes for 12 C/D box RNAs were localised of which 7 were predicted to modify rRNAs, 2 to target tRNAs and a further 2 to modify unknown RNAs. In addition, a single copy of a gene for an H/ACA box RNA was located which together with aPus7 should generate pseudouridine-35 in Sulfolobus pre-tRNATyr transcripts (Muller et al. 2009). However, in A. hospitalis, the aPus7 gene (Ahos0631) is degenerate. A further 10 genes were assigned to encode RNAs of unknown function. The relatively high conservation of sequence and gene synteny for these RNAs between Sulfolobus and Acidianus species underlines their potential functional importance.
Reading frame shifts and mRNA intron splicing
Examples of translational reading frame shifts yielding single polypeptides have been demonstrated experimentally for S. solfataricus P2 (Cobucci-Ponzano et al. 2010). For two of these, a transketolase (Ahos1219/1218) and a putative O-sialoglycoprotein endopeptidase (Ahos0695/0696), the A. hospitalis genes overlap in a similar way, and are likely to undergo translational frame shifts. Moreover, transcripts of the intron-carrying cbf5 gene (Ahos0734/0735) are likely to undergo splicing at the mRNA level by the archaeal splicing enzyme complex (Ahos0689/0798/1417) as has been demonstrated experimentally for different crenarchaea (Yokobori et al. 2009).
Metabolic pathways
Genome analyses indicate the presence of versatile metabolic pathways in A. hospitalis. They suggest that it can grow autotrophically by fixing CO2 or heterotrophically using yeast extract, as has been demonstrated experimentally (Basta et al. 2009). Genome analyses also revealed genes encoding sugar transporters and glycosidases suggesting that A. hospitalis can assimilate carbohydrates, such as starch, glucose, mannose and galactose. Moreover, enzymes are encoded that are implicated in energy generation from oxidising elemental sulphur, hydrogen sulphides and other reduced inorganic sulphide compounds, but not ferrous ions. However, no hydrogenase genes were detected suggesting that A. hospitalis cannot use H2 as electron donor for growth.
Enzymes were identified for a complete TCA cycle that is important for generating different intermediates for the biosynthesis of many cellular components, as well as producing reduced electron carriers, such as NAD(P)H, reduced ferredoxin (FdR) and FADH2. Formation of acetyl-CoA from pyruvate and the formation of succinyl-CoA from 2-oxoglutarate were predicted to be catalysed, respectively, by pyruvate ferredoxin oxidoreductase (Ahos1949-1952) and 2-oxoglutarate ferredoxin oxidoreductase (Ahos0089/0090/0300/0301). Moreover, both enzymes were predicted to use ferredoxin instead of NAD+ as a cofactor.
Genes encoding enzymes involved in pathways for fixing atmosphere N2, or reducing nitrate and nitrite, as nitrogen sources were absent, as observed for other Acidianus species, and the genome analyses suggest that ammonium is an exclusive source of nitrogen that is assimilated via formation of carbamoyl phosphate, glutamine and glutamate. Genes encoding putative carbamoyl phosphate synthetase (Ahos1106/1107), glutamine synthetase (Ahos0460, Ahos1272, Ahos2233) and glutamate dehydrogenase (Ahos0494) are present.
Sulphur metabolism
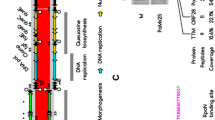
A. hospitalis encodes several enzymes involved in sulphur metabolism, including the oxidation and reduction of sulphur, the thiosulphate–tetrathionate cycle which generates sulphate, and the participation of sulphur in electron transport. However, genes for some sulphur metabolism enzymes, including sulphite-acceptor oxidoreductase, adenosine phosphosulphate reductase, sulphate adenylyl transferase and adenylylsulphate phosphate adenyltransferase were not found which suggested that A. hospitalis has some pathways differing from those of other Acidianus and Sulfolobus species (Kletzin 2007). Therefore, based on the gene annotations, a model is presented for the proposed sulphur oxidation and reduction pathways in A. hospitalis (Fig. 2). Extracellular H2S is oxidised by a secretory-type sulphide:quinone oxidoreductase (Ahos0513) and flavocytochrome c sulphide dehydrogenase (Ahos0188) to produce a surface layer of sulphur on the outer cell membrane. Elemental sulphur is then transported into the cell by putative-SH radical transporter(s) using an unknown mechanism. Subsequently, sulphur is oxidised by sulphur oxygenase-reductase (Ahos0131) to yield sulphite, thiosulphate and hydrogen sulphide. Sulphite and elemental sulphur convert spontaneously and non-enzymatically to thiosulphate and elemental sulphur and, consistent with this mechanism, no candidate gene encoding sulphite:acceptor oxidoreductase was identified in the A. hospitalis genome. Thiosulphate enters the putative thiosulphate/tetrathionate cycle and is finally oxidised to sulphate. The enzymes involved in this cycle were all annotated: thiosulphate:quinone oxidoreductase (Ahos0112-0113 and Ahos0238-0239) and tetrathionate hydrolase (Ahos1670). H2S is either oxidised by the sulphide:quinone oxidoreductase (Ahos1014) in the cytoplasm with quinone-cytochrome as electron acceptor or it reacts with tetrathionate spontaneously under the high temperature growth conditions. Finally, sulphate generated from sulphur oxidation is effluxed from the cell by a putative sulphate transport permease (Ahos1256). Electrons generated from sulphur oxidation enter the electron transport chain via quinone. Terminal quinol oxidase receives electrons from quinone and transfers them to O2 coupled with ATP generation. Some electrons may be transmitted to the NADH complex to produce NADH for use in other pathways.

Model of pathways for oxidation and reduction of sulphur in A. hospitalis indicating the predicted functions of genes in the A. hospitalis genome and corresponding gene numbers are given for each step. The following abbreviations are used: OM outer membrane, IM inner membrane, SQR sulphide:quinone oxidoreductase, Fcc flavocytochrome c sulphide dehydrogenase, SOR sulphur oxygenase-reductase, TetH tetrathionate hydrolase, TQO thiosulphate–quinone oxidoreductase; SulP sulphate transporter permease, QH 2 quinol pool
Transporters and proteolytic enzymes
Twenty-eight gene products were predicted to be involved in the transport of amino acids, oligopeptide/dipeptides and ammonium. Of these, 19 are implicated in amino acid transport, including 5 amino acid transporters (Ahos0100/0163/0197/0986/1721), three amino acid permeases (Ahos0328/0439/1725) and 11 amino acid permease-like proteins (Ahos0272/0276/0958/1040/1086/1868/1891/1907/1953/2065/2251) of unknown specificity for amino acid uptake. Genes encoding an ammonium transporter (Ahos1467) and two oligopeptide/dipeptide ABC transporter gene clusters (Ahos0337-0342 and Ahos0170-0175) are present. In addition, 21 genes were predicted to encode proteolytic enzymes, including 20 peptidases. Of these, four are endopeptidases (Ahos0428/0516/0695-6/0800), three are aminopeptidases (Ahos0013/0588/1941), two are pepsins (Ahos1929/2087) and one is a carboxypeptidase (Ahos0991). Five of the proteolytic enzymes are predicted to be membrane-bound and are designated secretory proteins. These results suggest that A. hospitalis, like Acidianus brierleyi (Segerer et al. 1986), Acidianus tengchongensis (He and Li 2004) and Acidianus manzaensis (Yoshida et al. 2006), can grow on organic compounds, such as yeast extract, peptone, tryptone and casamino acids.
Toxin–antitoxin systems
VapBC complexes constitute the main family of antitoxin–toxins that are encoded by members of the Sulfolobales (Pandey and Gerdes 2005; Guo et al. 2011), and they occur mainly in variable genomic regions where they may undergo loss or gain events (Guo et al. 2011). The A. hospitalis genome carries 26 vapBC gene pairs that are concentrated in the genomic regions 350–410 and 1,374–1,912 kb with a single vapC-like gene lying in an operon (Fig. 1). The VapB antitoxins, in contrast to VapC toxins, could be classified into three families of transcriptional regulators, AbrB, CcdA/CopG and DUF217 (Fig. 4a), whilst no subclassification was observed for the VapC proteins (Fig. 4b). Tree building based on the sequence alignments demonstrated that the sequences of these antitoxins and toxins are highly diverse, with sequence identities between them rarely exceeding 30%, as indicated by all the proteins exhibiting long branches (Fig. 4). This result contrasted with the finding that VapBC complexes with closely similar sequences are commonly found when comparing different genomes of the Sulfolobales. For example, 11 of the 26 VapBC protein pairs have closely similar homologs encoded in at least 7 of the 13 available Sulfolobus genomes (Fig. 4b). This indicates that there is likely to be a selection against the uptake of closely similar vapBC gene pairs in a given genome, despite the abundance of such gene pairs in the environment.
The A. hospitalis genome also encodes six copies of RelE-related toxin proteins, in common with other Sulfolobus genomes (Pandey and Gerdes 2005, unpublished results). At least three of the relE genes occur in integrated regions carrying degenerated conjugative plasmids, and they show sequence similarity to proteins encoded in Sulfolobus conjugative plasmids pKEF9 (ORF69b), pING1 (ORF98) and pL085 (gene no. 3195) (Greve et al. 2004; Stedman et al. 2000; Reno et al. 2009). However, none of the putative toxin genes are linked physically to antitoxin relB genes and their function remains unknown.
Diverse CRISPR-based immune systems
The CRISPR-based immune systems of A. hospitalis can be classified into two main types based on analyses of their Cas1 protein, leader and repeat sequences (Shah et al. 2009; Lillestøl et al. 2009). In total, there are six CRISPR loci, carrying 129 spacer-repeat units none of which are identical (Fig. 5). The first three loci in the genome (Ahos-53, -13 and -9a) are physically linked by cassettes of cmr and cas family genes, each of which contains a vapBC antitoxin–toxin gene pair, and they constitute a family II CRISPR/Cas system (Fig. 5a). The last two CRISPR loci (Ahos-9b and 5) are coupled into a typical family I paired CRISPR/Cas module (Fig. 5b) and there is a vapBC gene pair immediately upstream. Preceding the latter CRISPR/Cas module, there is a single unclassified locus (Ahos-40) that lacks both cas genes and a leader region (Fig. 5c) (Shah and Garrett 2011).
We analysed the degree to which CRISPR spacers exhibited sequence matches to the many diverse genetic elements available from Acidianus and Sulfolobus species using an earlier approach examining nucleotide and translated sequences of the spacers (Shah et al. 2009; Lillestøl et al. 2009). Relatively few significant sequence matches were found and most of these were to conjugative plasmids, with a few matches to members of five different viral families (Fig. 5).
Discussion
At about 2.1 Mbp, the genome of A. hospitalis is much smaller than other sequenced genomes of members of the Sulfolobales. Although this partly reflects the presence of low levels of transposable elements and few genes deriving from integrated elements, it also results from a lower diversity of metabolic and transporter genes (Guo et al. 2011). The Z curve analysis suggests that the chromosome carries three replication origins as for Sulfolobus species (Fig. 1), although in contrast to the sequenced strains of S. solfataricus and S. islandicus, the whiP/cdt1 and cdc6-2 genes are widely separated.
Although no systematic analysis has been performed experimentally on the metabolic capacity of A. hospitalis, genome analyses revealed that A. hospitalis possesses the capacity to assimilate a broad range of organic compounds, including different amino acids and proteolytic products, which is similar to some other Acidianus and Sulfolobus species (Segerer et al. 1986; Grogan 1989; He et al. 2004; Yoshida et al. 2006; Plumb et al. 2007). The analyses also support that A. hospitalis can assimilate various carbohydrates, similarly to several Sulfolobus species (Grogan 1989) but in contrast to some Acidianus species (Yoshida et al. 2006; Plumb et al. 2007).
A. hospitalis, like other Acidianus and Sulfolobus species, obtains energy for growth mainly via oxidation of reduced inorganic sulphuric components (RISCs), and the enzymes involved were predicted from the genome analyses (Fig. 2). A sulphur oxygenase-reductase was identified showing amino acid sequence similarity to other Acidianus and Sulfolobus SORs of 67–99%, and we inferred that it is important for elemental sulphur oxidation and reduction, as occurs in both Acidianus and Sulfolobus species (Kletzin 1989, 1992; Sun et al. 2003; Chen et al. 2005a). One product of sulphur oxygenase-reductase catalysis is sulphite. Owing to the apparent lack of the four enzymes, sulphite-acceptor oxidoreductase, adenosine phosphosulphate reductase, sulphate adenylyl transferase and adenylylsulphate phosphate adenyltransferase, A. hospitalis must have adopted a strategy for sulphite oxidation that differs from the currently known pathway (Kletzin 2007). Here, we propose that sulphite is channelled to thiosulphate in A. hospitalis via a spontaneous reaction with elemental sulphur, but this remains to be tested experimentally. Some Acidianus species, such as A. manzaensis (Yoshida et al. 2006) and A. sulfidivorans (Plumb et al. 2007) grow chemolithoautotrophically with oxidation of molecular hydrogen, but this cannot occur in A. hospitalis because it apparently lacks an encoded hydrogen dehydrogenase.
Transposable elements include a few IS200/607 elements and several orphan orfB elements which all belong to the IS200/605/607 family. They lack inverted terminal repeats and are mobilised by “cut-and-paste” mechanisms (Filée et al. 2007; Ton-Hoang et al. 2010). No representatives of other transposable element families were found, common to other Sulfolobus genomes, which carry inverted terminal repeats and are mobilised by “copy-and-paste” mechanisms (Blount and Grogan 2005; Redder and Garrett 2006). It remains uncertain whether the OrfB protein is responsible for transposition of the orfB elements or whether they are mobilised in trans by the TnpA transposase encoded by the IS200/607 elements (Filée et al. 2007; Guo et al. 2011). The IS200/607 and orfB elements have been detected in Sulfolobus conjugative plasmids and orfB elements also occur in a few viruses of the Sulfolobales including four copies in the Acidianus two-tailed bicaudavirus ATV (She et al. 1998; Greve et al. 2004; Prangishvili et al. 2006). Thus, they are likely to be transmitted intercellularly, and enter chromosomes, via such genetic elements.
MITEs are common in Sulfolobus species and have been predicted to be mobilised by transposases encoded in different IS element families (Redder et al. 2001). The novel MITE-like elements in the A. hospitalis genome (Fig. 3) may derive from orfB elements and be mobilised by a similar mechanism but at present we can provide no evidence for their mobility. In this respect, they may be similar to other Sulfolobus MITEs which show a low level of transpositional activity (Redder and Garrett 2006). This is consistent with the hypothesis that MITEs drive the evolutionary diversification of their mobilising transposases to the point that they are no longer recognised which leads to their immobilisation and subsequent degeneration (Feschotte and Pritham 2007).

Alignment of 10 MITE-like repeat elements present in the genome of A. hospitalis. The shaded area denotes to a small open reading frame corresponding to the downstream part of the OrfB found within transposable orfB elements
All of the integrated elements, except one, could be identified as originating from fuselloviruses or a pDL10-like member of the pRN family of cryptic plasmids (Kletzin et al. 1999), and the conjugative plasmid pAH1 was already shown to reversibly integrate at a tRNAArg [TCG] gene (Basta et al. 2009). None of these events occurred within any of the 15 tRNA genes carrying introns and this observation is consistent with the hypothesis that archaeal introns protect tRNA genes against integration events (Guo et al. 2011).
VapBC constitutes the predominant antitoxin–toxin family found amongst the Sulfolobales and the A. hospitalis genome carries 26 vapBC gene pairs, more than occur in more rapidly growing Sulfolobus species (Pandey and Gerdes 2005; Guo et al. 2011). Moreover, the groups of VapB and VapC proteins are highly diverse in sequence (Fig. 4). Antitoxin–toxins were originally shown to enhance plasmid maintenance as a consequence of the growth of plasmid-free cells being preferentially inhibited by free toxins which are inherently more stable than the antitoxins (Gerdes 2000). By analogy with this mechanism, it was proposed that chromosomally encoded toxins may facilitate maintenance of local DNA regions where vapBC gene pairs are located that might otherwise be prone to loss (Magnuson 2007; Van Melderen 2010). This hypothesis is consistent with the observation that most of the A. hospitalis vapBC gene pairs lie within the two variable genomic regions where DNA regions are exchanged (Fig. 1). Moreover, it receives strong support from both the high diversity, and the uniqueness of all the VapC proteins encoded within the A. hospitalis chromosome (Fig. 4b), because any similar VapBC complexes would compensate for the loss of one another, thereby undermining any DNA maintenance activity.

VapBC trees. Phylogenetic trees for a VapB antitoxins and b VapC toxins. They demonstrate that VapBs, despite their high sequence diversity, can be classified into three main families AbrB, CcdA/CopG and DUF217, whereas the VapCs are highly diverse in their sequences but cannot be classified into major subgroups. The Ahos gene numbers are given for each protein. Moreover, the class of the VapB corresponding to each VapC is given in b. The degree of conservation of the VapC proteins in the available 13 Sulfolobus genomes is indicated in b where 0 indicates it is absent from all the genomes whilst 13 indicates that it is present in all. The antitoxin corresponding to VapC-0183 is not annotated in the genome because it lacks a start codon but it is included in the figure. The VapC-like protein (Ahos0712) is part of the operon with a translation-related protein and lacks a VapB. The Ahos1664/1663 pair are variant ORFs where both VapB and VapC are longer than usual and the VapB does not cluster with the families in a
In slowly growing organisms, from nutrient poor environments, multiple toxins are also assumed to be involved in stress response and/or quality control (Gerdes 2000; Pandey and Gerdes 2005). Involvement in stress response entails that the more stable toxins inhibit growth and allow the host to lie in a dormant state during the period of environmental stress (Gerdes 2000). However, there may also be a negative effect on host growth due to the continuous presence of low levels of free toxin (Wilbur et al. 2005). Thus, the presence of many vapBC gene pairs in A. hospitalis could reflect a compromise between the ability to survive different environmental stresses and maintaining an adequate growth rate under normal conditions. This would be also consistent with the presence of three families of VapB proteins and high sequence diversity of the VapC proteins, since functionally overlapping systems would be redundant for stress responses and they would confer an unnecessary burden on host growth. The proposed dual roles of maintenance of local chromosomal DNA regions and providing resistance to stress and are not mutually exclusive.
Although the mechanism of action of VapC toxins remains unknown (Arcus et al. 2011), in A. hospitalis, a single vapC-like gene (Ahos0712) is directly coupled to genes encoding proteins involved in transcription and initiator tRNA binding to the ribosome, and this gene cassette is highly conserved in gene content and sequence in other Sulfolobus genomes (Guo et al. 2011). This suggests that this VapC protein, at least, may also regulate or inhibit translational initiation by binding at the ribosomal A-site, as demonstrated recently for a RelE type toxin (Neubauer et al. 2009). A similar inactivation mechanism would be plausible for the VapC toxins, if one assumes that expression of the individual VapBC complexes is stimulated by either the requirement to maintain different local regions of chromosomal DNA or different environmental stresses.
Despite the complexity of the CRISPR-based immune systems present in the genome, they appear to be, at best, only partially functional. Thus, the family II CRISPR/Cas system is coupled with an archaeal family D Cmr module in A. hospitalis, but is apparently defective, retaining only its putative RNA, but not DNA, targeting function. The system lacks the group 2 cas genes (cas3, cas5, csa2, csa5, csaX) which encode proteins implicated in targeting and inactivating foreign DNA elements (Fig. 5). However, the cas group 1 genes (cas1, cas2, cas4, csa1), putatively involved in integrating new spacers from invading DNA elements are present, and the Cmr module implicated in RNA targeting are also present (Garrett et al. 2011; Shah et al. 2011). The family I system exhibits small CRISPR loci, with intact leader regions and group 2 cas genes. However, the cas2 gene in the group 1 cas gene cassette is truncated, having incurred a point mutation which produces a premature stop codon. Thus, this system has apparently lost the ability to integrate new spacers. This suggests that neither CRISPR-based system is fully functional, despite their apparent complexity. The presence of five vapBC gene pairs located either within the cmr and cas gene cassettes of the family II CRISPR/Cas module, or immediately upstream from the modules of both families, may reflect that they help to maintain these gene cassettes on the chromosome (see above).

Schematic representations of the CRISPR loci of A. hospitalis. a Family II CRISPR module carrying three CRISPR loci and Cmr and Cas family gene cassettes which are both interrupted by, or bordered by, four vapBC gene pairs (orange). b Paired family I CRISPR/Cas system flanked by one vapBC gene pair, and c. an unclassified CRISPR locus lacking a leader region and adjacent cas genes. csm1 is a homolog of cmr2, csm2 is a homolog of cmr5 and csm3 is a homolog of cmr4. The light blue genes each carry two short RAMP motifs. a–c Structures of the individual CRISPR loci are shown together with the leader region (L) where each triangle represents a spacer-repeat unit. Significant spacer matches to sequenced viruses and plasmids are colour coded: red rudivirus, orange lipothrixvirus, yellow fusellovirus, green bicaudavirus, turquoise turreted icosahedral virus, blue conjugative plasmid and violet cryptic plasmid
Although a range of genetic systems have been developed for Sulfolobus species, at present no genetic systems are available for the Acidianus genus and A. hospitalis provides a promising candidate for such studies. It has a minimal size and the relative stability of its chromosome suggests that it is likely to generate stable deletion mutants. This, combined with its ability to host different plasmids and viruses provides a promising starting point for developing a genetic system.
References
Arcus VL, McKenzie JL, Robson J, Cook GM (2011) The PIN-domain ribonucleases and the prokaryotic VapBC toxin–antitoxin array. Prot Engin Design Select 24:33–40
Basta T, Smyth J, Forterre P, Prangishvili D, Peng X (2009) Novel archaeal plasmid pAH1 and its interactions with the lipothrixvirus AFV1. Mol Microbiol 71:23–34
Bettstetter M, Peng X, Garrett RA, Prangishvili D (2003) AFV1, a novel virus infecting hyperthermophilic archaea of the genus Acidianus. Virology 315:68–79
Blount ZD, Grogan DW (2005) New insertion sequences of Sulfolobus: functional properties and implications for genome evolution in hyperthermophilic archaea. Mol Microbiol 55:312–325
Chen Z-W, Jiang C-Y, She Q, Liu S-J, Zhou P-J (2005a) Key role of cysteine residues in catalysis and subcellular localization of sulfur oxygenase reductase of Acidianus tengchongensis. Appl Environ Microbiol 71:621–628
Chen L, Brügger K, Skovgaard M, Redder P, She Q, Torarinsson E, Greve B, Awayez M, Zibat A, Klenk HP, Garrett RA (2005b) The genome of Sulfolobus acidocaldarius, a model organism of the Crenarchaeota. J Bacteriol 187:4992–4999
Cobucci-Ponzano B, Guzzini L, Benelli D, Londei P, Perrodou E, Lecompte O, Tran D, Sun J, Wei J, Mathur EJ, Rossi M, Moracci M (2010) Functional characterisation and high-throughput proteomic analysis of interrupted genes in the archaeon Sulfolobus solfataricus. J Proteome Res 9:2496–2507
Feschotte C, Pritham EJ (2007) DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet 41:331–368
Filée J, Siguier P, Chandler M (2007) Insertion sequence diversity in archaea. Microbiol Mol Biol Revs 71:121–157
Garrett RA, Shah SA, Vestergaard G, Deng L, Gudbergsdottir S, Kenchappa CS, Erdmann S, She Q (2011) CRISPR-based immune systems of the Sulfolobales—complexity and diversity. Biochem Soc Trans 39:51–57
Gerdes K (2000) Toxin-antitoxin modules may regulate dynthsis of macromolecules during nutritional stress. J Bacteriol 182:561–572
Goulet A, Blangy S, Redder P, Prangishvili D, Felisberto-Rodrigues C, Forterre P, Campanacci V, Cambillau C (2009) Acidianus filamentous virus 1 coat proteins display a helical fold spanning the filamentous archaeal viruses lineage. Proc Natl Acad Sci USA 106:21155–21160
Greve B, Jensen S, Brügger K, Zillig W, Garrett RA (2004) Genomic comparison of archaeal conjugative plasmids from Sulfolobus. Archaea 1:231–239
Grogan DW (1989) Phenotypic characterization of the archaebacterial genus Sulfolobus: comparison of five wild-type strains. J Bacteriol 171:6710–6719
Guo L, Brügger K, Liu C, Shah SA, Zheng H, Zhu Y, Wang S, Lillestøl RK, Chen L, Frank J, Prangishvili D, Paulin L, She Q, Huang L, Garrett RA (2011) Genome analyses of Icelandic strains of Sulfolobus islandicus: model organisms for genetic and virus-host interaction studies. J Bacteriol 193:1672–1680
He Z-G, Zhong H, Li Y (2004) Acidianus tengchongensis sp. nov., a new species of acidothermophilic archaeon isolated from an acidothermal spring. Curr Microbiol 48:156–193
Kletzin A (1989) Coupled enzymatic production of sulfite, thiosulfate, and hydrogen sulfide from sulfur: purification and properties of a sulfur oxygenase reductase from the facultatively anaerobic archaebacterium Desulfurolobus ambivalens. J Bacteriol 171:1638–1643
Kletzin A (1992) Molecular characterization of the sor gene, which encodes the sulfur oxygenase/reductase of the thermoacidophilic Archaeum Desulfurolobus ambivalens. J Bacteriol 174:5854–5859
Kletzin A (2007) Oxidation of sulfur and inorganic sulfur compounds in Acidianus ambivalens. In: Dahl C, Friedrich CG (eds) Microbial sulfur metabolism. Springer, Heidelberg, pp 184–199
Kletzin A, Lieke A, Urich T, Charlebois RL, Sensen CW (1999) Molecular analysis of pDL10 from Acidianus ambivalens reveals a family of related plasmids from extremely thermophilic and acidophilic archaea. Genetics 152:1307–1314
Lawrence CM, Menon S, Eilers BJ, Bothner B, Khayat R, Douglas T, Young MJ (2009) Structural and functional studies of archaeal viruses. J Biol Chem 284:12599–12603
Lillestøl RK, Shah SA, Brügger K, Redder P, Phan H, Christiansen J, Garrett RA (2009) CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol 72:259–272
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Lundgren M, Andersson A, Chen L, Nilsson P, Bernander R (2004) Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc Natl Acad Sci USA 101:7046–7051
Magnuson RD (2007) Hypothetical functions of toxin–antitoxin systems. J Bacteriol 189:6089–6092
Melderen LV (2010) Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol 13:781–785
Muller S, Urban A, Hecker A, Leclerc A, Branlant C, Motorin Y (2009) Deficiency of the tRNATyr:Ψ35-synthase aPus7 in archaea of the Sulfolobales order might be rescued by the H/ACA sRNA-guided machinery. Nucleic Acids Res 37:1308–1322
Muskhelishvili G, Palm P, Zillig W (1993) SSV1-encoded site-specific recombination system in Sulfolobus shibatae. Mol Gen Genet 273:334–342
Neubauer C, Gao YG, Andersen KR, Dunham CM, Kelley AC, Hentschel J, Gerdes K, Ramakrishnan V, Brodersen DE (2009) The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell 139:1084–1095
Omer AD, Zago M, Chang A, Dennis PP (2006) Probing the structure and function of an archaeal C/D-box methylation guide sRNA. RNA 12:1708–1720
Pandey DP, Gerdes K (2005) Toxin-antitoxin loci are highly abundant in free-living but lost from host-asscoiated prokaryotes. Nucleic Acids Res 33:966–976
Plumb JJ, Haddad CM, Gibson JAE, Franzmann PD (2007) Acidianus sulfidivorans sp nov., an extremely acidophilic, thermophilic archaeon isolated from a solfatara on Lihir Island, Papua New Guinea, and amendation of the genus description. Int J Syst Evol Microbiol 57:1418–1423
Prangishvili D, Albers SV, Holz I, Arnold HP, Stedman K, Klein T, Singh H, Hiort J, Schweier A, Kristjansson JK, Zillig W (1998) Conjugation in archaea: frequent occurrence of conjugative plasmids in Sulfolobus. Plasmid 40:190–202
Prangishvili D, Forterre P, Garrett RA (2006) Viruses of the Archaea: a unifying view. Nat Rev Microbiol 4:837–848
Rachel R, Bettstetter M, Hedlund BP, Häring M, Kessler A, Stetter KO, Prangishvili D (2002) Arch Virol 147:2419–2429
Redder P, Garrett RA (2006) Mutations and rearrangements in the genome of Sulfolobus solfataricus P2. J Bacteriol 188:4198–4206
Redder P, She Q, Garrett RA (2001) Non-autonomous elements in the crenarchaeon Sulfolobus solfataricus. J Mol Biol 306:1–6
Redder P, Peng X, Brügger K, Shah SA, Roesch F, Greve B, She Q, Schleper C, Forterre P, Garrett RA, Prangishvili D (2009) Four newly isolated fuselloviruses from extreme geothermal environments reveal unusual morphologies and a possible interviral recombination mechanism. Environ Microbiol 11:2849–2862
Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ (2009) Sulfolobus islandicus pan-genome. Proc Natl Acad Sci USA 106:8605–8610
Robinson NP, Bell SD (2007) Extrachromosomal element capture and the evolution of multiple replication origins in archaeal chromosomes. Proc Natl Acad Sci USA 104:5806–5811
Robinson NP, Dionne I, Lundgren M, Marsh VL, Bernander R, Bell SD (2004) Identification of two origins of replication in the single chromosome of the archaeon Sulfolobus solfataricus. Cell 116:25–38
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945
Segerer A, Neuner A, Kristjansson JK, Stetter KO (1986) Acidanus infernus gen. nov., sp. nov., and Acidianus brierleyi comb. nov.: facultatively aerobic, extremely acidophilic thermophilic sulfur-metabolizing archaebacteria. Int J Syst Bacteriol 36:559–564
Shah SA, Garrett RA (2011) CRISPR/Cas and Cmr modules, mobility and evolution of adaptive immune systems. Res Microbiol 162:27–38
Shah SA, Hansen NR, Garrett RA (2009) Distributions of CRISPR spacer matches in viruses and plasmids of crenarchaeal acidothermophiles and implications for their inhibitory mechanism. Trans Biochem Soc 37:23–28
Shah SA, Vestergaard G, Garrett RA (2011) CRISPR/Cas and CRISPR/Cmr immune systems of archaea. In: Marchfelder A, Hess W (eds) Regulatory RNAs in prokaryotes. Springer, Berlin
She Q, Phan H, Garrett RA, Albers S-V, Stedman KM, Zillig W (1998) Genetic profile of pNOB8 from Sulfolobus: the first conjugative plasmid from an archaeon. Extremophiles 2:417–425
Stedman KM, She Q, Phan H, Holz I, Singh H, Prangishvili D, Garrett RA, Zillig W (2000) The pING family of conjugative plasmids from the extremely thermophilic archaeon Sulfolobus islandicus: insights into recombination and conjugation in Crenarchaeota. J Bacteriol 182:7014–7020
Sun CW, Chen ZW, He ZG, Zhou PJ, Liu SJ (2003) Purification and properties of the sulphur oxygenase/reductase from the acidothermophilic archaeon, Acidianus strain S5. Extremophiles 7:131–134
Tang TH, Polacek N, Zywicki M, Huber H, Brügger K, Garrett R, Bachellerie JP, Hüttenhofer A (2005) Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol Microbiol 55:469–481
Ton-Hoang B, Pasternak C, Siguier P, Guynet C, Hickman AB, Dyda F, Sommer S, Chandler M (2010) Single-stranded DNA transposition is coupled to host replication. Cell 142:398–408
Torarinsson E, Klenk H-P, Garrett RA (2005) Divergent transcriptional and translational signals in Archaea. Environ Microbiol 7:47–54
Wilbur JS, Chivers PT, Mattison K, Potter L, Brennan RG, So M (2005) Neisseria gonorrheae FitA interacts with FitB to bind DNA through its ribbon–helix–helix motif. Biochemistry 44:12515–12524
Wurtzel O, Sapra R, Chen F, Zhu ZY, Simmons BA, Sorek R (2010) A single-base resolution map of an archaeal transcriptome. Genome Res 20:133–141
Yokobori S, Itoh T, Yoshinari S, Nomura N, Sako Y, Yamagishi A, Oshima T, Kita K, Watanabe Y (2009) Gain and loss of an intron in a protein-coding gene in Archaea: the case of an archaeal RNA pseudouridine synthase gene. BMC Evol Biol 9:198
Yoshida N, Nakasato M, Ohmura N, Ando A, Saolo J, Ishii M, Igarashi Y (2006) Acidianus manzaensis sp. nov., a novel thermoacidophilic Archaeon growing autotrophicallly by the oxidation of H2 with the reduction of Fe3+. Curr Microbiol 53:406–411
Zhang R, Zhang CT (2003) Multiple replication origins of the archaeon Halobacterium species NRC-1. Biochem Biophys Res Comm 302:728–734
Acknowledgments
We thank Mery Pina and Tamara Basta for help with the DNA preparation. The work was supported by the National Nature Science Foundation of China (30621005) and the Ministry of Science and Technology (2010CB630903), and by the Danish Natural Science Research Council (Grant no. 272-08-0391) and Danish National Research Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by L. Huang.
X.-Y. You and C. Liu contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
You, XY., Liu, C., Wang, SY. et al. Genomic analysis of Acidianus hospitalis W1 a host for studying crenarchaeal virus and plasmid life cycles. Extremophiles 15, 487–497 (2011). https://doi.org/10.1007/s00792-011-0379-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-011-0379-y