
Abstract
Here, we combined flow cytometry (FCM) and phylogenetic analyses after cell sorting to characterize the dominant groups of the prokaryotic assemblages inhabiting two ponds of increasing salinity: a crystallizer pond (TS) with a salinity of 390 g/L, and the non-crystallizer pond (M1) with a salinity of 200 g/L retrieved from the solar saltern of Sfax in Tunisia. As expected, FCM analysis enabled the resolution of high nucleic acid content (HNA) and low nucleic acid content (LNA) prokaryotes. Next, we performed a taxonomic analysis of the bacterial and archaeal communities comprising the two most populated clusters by phylogenetic analyses of 16S rRNA gene clone library. We show for the first time that the presence of HNA and LNA content cells could also be extended to the archaeal populations. Archaea were detected in all M1 and TS samples, whereas representatives of Bacteria were detected only in LNA for M1 and HNA for TS. Although most of the archaeal sequences remained undetermined, other clones were most frequently affiliated to Haloquadratum and Halorubrum. In contrast, most bacterial clones belonged to the Alphaproteobacteria class (Phyllobacterium genus) in M1 samples and to the Bacteroidetes phylum (Sphingobacteria and Salinibacter genus) in TS samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coastal solar salterns, characterized by extreme salt concentrations, are colonized by a variety of extreme halophilic–heterotrophic prokaryotes (Oren 2002a). Knowing the diversity and activity of halophilic prokaryotes is therefore of fundamental importance to understand their contribution to organic matter recycling and their functional role in particular environments (Pedrós-Alió et al. 2000; Elloumi et al. 2009).
Culture-dependent methods were shown insufficient to describe bacterial and archaeal diversity from natural samples (Hugenholtz et al. 1998). The use of 16S rRNA clone libraries has led to the discovery of many novel microbial taxa, leading to a better knowledge of the diversity of prokaryotic communities inhabiting extreme salt environments (Benlloch et al. 2002). Comparison of 16S rRNA clone libraries obtained from several extreme salt environments from different regions of the world has revealed unexpected uniformity among the microbial communities (Oren 2002a). For instance, it was reported that crystallizers from Spain, Israel, Australia, Tunisia and Turkey with salinities approaching saturation are dominated by the square archaeon related to the recently isolated Haloquadratum walsbyi (Burns et al. 2007; Benlloch et al. 2001, 2002; Antón et al. 2000; Oren 2002b; Ochsenreiter et al. 2002; Baati et al. 2008; Maturrano et al. 2006). Salinibacter ruber, a representative of the Bacteroidetes phylum of Bacteria, is also a common inhabitant of extreme salt environments worldwide which are at or approaching halite saturation (Antón et al. 2002; Oren and Rodríguez-Valera 2001; Elevi Bardavid et al. 2007; Maturrano et al. 2006; Burns et al. 2007; Jiang et al. 2006). However, some different assemblages were also reported, such as those characterized by the presence of many Halobacterium- and Salicola-related phylotypes in Andean salterns (Maturrano et al. 2006) or the dominance of Halorubrum, Haloarcula or Halobacterium spp. in extreme salt ponds where Haloquadratum are not dominant (Pasić et al. 2007; Bowman et al. 2000; Bidle et al. 2005). Salinibacter, Haloquadratum, and Halorubrum genera have been recently reported to be abundant in a crystallizer pond of solar saltern of Sfax in Tunisia (Baati et al. 2008).
In the last two decades, flow cytometry (FCM) has become one of the most powerful alternatives to microbial counts by optical microscopy in aquatic environments (Gasol and del Giorgio 2000; Grégori et al. 2001; Marie et al. 2000; Legendre et al. 2001; Pan et al. 2005; Casamayor et al. 2007). Besides determining abundance, FCM provides cellular information from individual cells; the forward angle light scatter is related to cell size, and the right angle light scatter is related to cell shape and structure. Fluorescence induced by particular dyes can also provide information on the heterogeneity in cell structure or physiology. For instance, the nucleic acid content of single cells can be assessed after specific staining with nucleic acid dyes (Button et al. 1996; Button and Robertson 2001). One of the most important findings from FCM studies is that aquatic Bacteria usually tend to cluster into at least two distinct cell fractions. One fraction is characterized by high fluorescence intensity, indicating high nucleic acid content (HNA). The second fraction is characterized by lower fluorescence intensity, indicating low nucleic acid content (LNA) (Button et al. 1996; Gasol et al. 1999; Troussellier et al. 1999; Lebaron et al. 2001; Nishimura et al. 2005; Sherr et al. 2006). Numerous studies have focused on the relative abundance of LNA and HNA Bacteria in aquatic environments (freshwater and seawater), in relation with their in situ activities and phylogenetic diversity (Jellett et al. 1996; Marie et al. 1997; Zubkov et al. 2001; Longnecker et al. 2005; Wang et al. 2009; Nishimura et al. 2005). To date, phylogenetic identification of the sorted HNA and LNA bacterial groups has been based on CARD-FISH (Zubkov et al. 2002) or DGGE analysis (Longnecker et al. 2005). Differences in the phylogenetic diversities of HNA and LNA cells have been reported for the community of Bacteria of the Northern Sea (Zubkov et al. 2001, 2002) and the Oregon coast (Longnecker et al. 2005), although samples from the Mediterranean sea revealed that both groups were composed of the same dominant species (Servais et al. 2003).
To our knowledge, combining FCM and phylogenetic analysis based on construction of 16S rRNA gene library, after cell sorting, has never been used to characterize heterotrophic microbial assemblages in extreme salt water. Moreover, the broad classification of the LNA and HNA fractions has never been assigned to archaeal community. The present study thus aimed to discriminate HNA and LNA cell groups (Bacteria and Archaea) with FCM and use phylogenetic analysis, after cell sorting, for unveiling new basic information on the distribution of heterotrophic prokaryotes inhabiting crystallizer and non-crystallizer ponds of coastal solar salterns.
Materials and methods
Sampling site and water collection
The study was conducted in the multipond solar saltern of Sfax (central-eastern coast of Tunisia, about 34°39′N and 10°42′E). The area of about 1500 ha is divided into 6 different series of ponds along the 12 km coastline. These ponds are shallow (20–70 cm deep), with salinity ranging between 40 and 400 g/L, the extreme salt conditions being favored by the arid climate. The process begins by storing seawater in 17 primary ponds, and water salinity is increased by evaporation. When the salt concentration reaches the range of 41–75 g/L, the seawater is moved to an internal part made of 5 parallel water ponds, until they reach a salt concentration of 130 g/L. After this stage, the seawater is distributed to the 6 pre-crystallization ponds to attain a salinity of 300 g/L. At the final stage in the crystallizer ponds, where the salt precipitates, brines reach a very high salt concentration (400–430 g/L; for more details, refer to Ayadi et al. 2002; Elloumi et al. 2009; Fig. 1). Throughout the present study, we used two ponds: a crystallizer (TS) with a salinity of about 390 g/L and a non-crystallizer (M1) with a salinity of about 200 g/L. From each sampling site, three water samples were collected on October 2009. One liter sample, kept at 4°C, was used for physico-chemical analyses and total cell counts. This was processed within 2 h after being sampled, while 4.5 mL was immediately fixed with 0.5 mL of a 20% (w/v) borate-buffered formalin solution, pH 7.2, and then rapidly frozen in liquid nitrogen and stored at −80°C until analysis by FCM.

Map of the location of the sampling ponds (M1) and (TS) of the Sfax Solar Saltern in Tunisia (Amdouni 1990)
Physico-chemical analyses
The following environmental parameters were measured in situ at the time of sampling: water temperature with a mercury glass thermometer, electrical conductivity (EC) with a probe conductivimeter (model Horiba U. 10), and dissolved oxygen concentration with an oxymeter probe (YSI 57). pH was measured by a Methrom type pH meter just after arrival at the laboratory. Salt concentration was determined at 120°C by drying 50 mL sample in a previously sterilized crystallizing dish, and calculating the total salt concentration from the difference in weight before and after evaporation. Samples for dissolved organic carbon (DOC) were filtered through 0.22 μm pore size membrane and the concentrations were measured as CO2 generated by catalytic combustion using a Shimadzu TOC-V carbon analyzer. The nitrogen content was assayed using the Kjeldahl method.
Heterotrophic prokaryote cell groups and total cell counts by FCM
FCM analyses for heterotrophic prokaryotes were conducted at the Regional Flow Cytometry Platform for Microbiology (PRECYM) at the Oceanology Centre of Marseille (http://precym.com.univ-mrs.fr) using a cell sorter flow cytometer (MoFlo™, Beckman-Coulter), equipped with a dual line, water-cooled, Coherent™ argon laser (351 and 488 nm). The MoFlo flow rate was determined by weighing a sample before and after 3 min of uptake by the instrument, the procedure being repeated 3 times. The average mass going into the instrument over 3 min was then converted into volume by dividing it by the mass of 100 μL of sample. The flow rate expressed in μL/s was derived from the average volume uptake during 3 min.
The extreme salt samples were thawed at room temperature, homogenized and a subsample of 0.5 mL was incubated for 15 min in the dark with 5 μL of SYBRGreen solution (Sigma, Germany, commercial solution diluted 1000 times, final concentration) to stain the nucleic acids. The SYBRGreen family dyes are widely used to stain nucleic acids of aquatic bacteria and in particular marine bacteria because of its weak sensitivity to salt (Lebaron et al. 1998). In addition, SYBRGreen was a dye of choice because of its high fluorescence yield when bound to nucleic acids (http://www.invitrogen.com).
Each cell was characterized by three optical parameters: forward (FSC) and right angle light scatter (SSC) of the photons from the 488 nm laser beam, and SYBRGreen fluorescence induced by the 488 nm excitation beam and collected in the green range 515–530 nm. Just before analysis, 10 μL of 1 μm fluorescent fluorosphere suspension (FluoresbryteTM, Polysciences) was added to normalize and control the flow cytometer settings. Data were collected and stored in list mode with the Summit software package (DAKO). Cell groups were optically resolved on the SSC versus SYBRGreen fluorescence and SSC versus FS plots by manual gating (drawing a region around each group). Combining SYBRGreen fluorescence and light diffusion parameters helps to distinguish unambiguously the cells from inorganic particles, detritus and free DNA (Marie et al. 2000).
The contribution percentage of each subgroup to the prokaryotic community was determined by dividing their abundance by the overall abundance of the analyzed prokaryotic cells. The FCM analysis and sorting were optimized by (a) running the sample at <8000 events s−1 in order to avoid coincidences and aborts (linked to the 5.5 μs dead time needed for data acquisition and transfer) and (b) gating out doublets based on the pulse width of the green fluorescence signal (SYBRGreen) used to trigger data acquisition.
Cell sorting based on size classes and nucleic acid content for molecular analysis
Preliminary assays showed that molecular analyses required at least 106 cells to obtain a good PCR amplification profile (data not shown). For molecular analysis, only two clusters (LNA and HNA) from the crystalliser (TS) and non-crystalliser (M1) ponds were considered for cell sorting. The cell abundance (<5%) in the other clusters was considered less favorable. The sorted cells were, therefore, simultaneously counted and collected in MilliQ water for phylogenetic analyses. The purity of the sorted groups was controlled by reanalyzing subsamples after each sorting.
PCR amplification of 16S rRNA genes, cloning and sequencing
Phylogenetic analyses were carried out on subgroups differentiated by FCM at both M1 and TS sites. Immediately after cell sorting, samples were heated at 98°C for 10 min then frozen at −80°C for 10 min to release the DNA. The sample obtained after the thermic shock was used as a template for PCR amplification of 16S with specific bacterial 16S rRNA gene primers (Weisburg et al. 1991), fD1 (5′-AGAGTTTGATCCTGGCTCAG-3′, corresponding to positions 7–26 in the Escherichia coli 16S rRNA gene) and rP2 (5′-ACGGCTACCTTGTTACGACTT-3′, corresponding to positions 1513–1494) and specific archaeal 16S rRNA gene primers (DeLong 1992), 21f (5′-TTCCGGTTGATCCYGCCGGA-3′) and 958r (5′-YCCGGCGTTGAMTCCAATT-3′). Each amplification reaction mixture (50 μL) contained PCR buffer (1×), 0.2 mM of each dNTP, 0.1 μM of each primer, 2 μL of template DNA and 1 U of High Fidelity expand DNA polymerase (Roche). After initial denaturation (94°C for 5 min), 45 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 1 min were performed, followed by a final extension (10 min, 72°C). The PCR products were purified, ligated to pGEM T-easy vector (Promega) and transformed into E. coli DH5α cells. Single colonies containing inserts were selected at random, and the inserts were amplified from cells using the primers Sp6 (5′-ATTTAGGTGACACTATAGAATAC-3′) and T7 (5′-GTAATACGACTCACTATAGGGC-3′). Successful transformants were inoculated in microplates containing 150 μL Luria-Broth medium supplemented with 40% glycerol and ampicillin (50 μg mL−1). Plasmid extraction and 16S rRNA gene sequencing were performed by the Cogenics service (http://www.cogenicsonline.com).
The 16S rRNA gene sequences were registered in the GenBank Data Library (http://www.ncbi.nlm.nih.gov) under accession numbers HQ157568 to HQ157683.
Phylogenetic analyses
The diversity of 16S rRNA gene sequences in the collected samples was investigated using the classifier tool (version 2.2) of the Ribosomal Database Project (RDP) website (http://rdp.cme.msu.edu/index.jsp; Cole et al. 2009). Only taxonomic affiliations with a confidence threshold of at least 95% were considered.
The taxonomic affiliations proposed by the classifier tool were verified by phylogenetic analysis. Closest homologs of each of the 16S rRNA gene sequences were identified by blastN in the nr database at the NCBI (Altschul et al. 1997) and retrieved. Our 16S rRNA sequences and their homologues retrieved from the nr database were aligned using clustalw2 and default parameters (Larkin et al. 2007). The resulting alignments were visually inspected and manually refined when necessary using the MUST package (Philippe 1993). Regions of the alignment where homology between sites was doubtful were removed. Bayesian phylogenetic trees were constructed using MrBayes 3.2 (Ronquist and Huelsenbeck 2003). We used a mixed model of nucleic acids substitution and a Γ law (four discrete categories plus a proportion of invariant sites) to account for among-site rate variation. The MrBayes software was run with four chains for 1 million generations and trees were sampled every 100 generations. To construct the consensus tree, the first 1500 trees were discarded as “burnin”.
Results
Physico-chemical analysis of water samples
The multipond of the Sfax solar saltern in Tunisia consists of a series of interconnected ponds with increasing degrees of salinity from seawater up to salt saturation. In this study, we focused our interest on two ponds with a large difference in salt concentration: the concentrator pond M1 (200 g/L) and the crystallizer pond TS (390 g/L) (Fig. 1). The physico-chemical characteristics of the sampling-sites surface waters are shown in Table 1.
Cytometric characterization of HNA and LNA cell groups in M1 and TS
FCM analysis of the collected samples at both sampling sites showed that the prokaryote abundance was about 3-fold higher in TS than in M1: 28.7 × 107 and 8.12 × 107 cells/mL, respectively. The resolution of distinct cell groups was achieved on the basis of their optical properties (forward and right angle light scatter, and SYBRGreen induced fluorescence) as illustrated in Fig. 2. The HNA and LNA clusters were observed at both M1 and TS sites and represented more than 95% of the total cell count, the cell abundances of the other clusters remaining <5%. In addition, LNA and HNA groups were partially overlapping and therefore not fully resolved on the basis of green fluorescence alone. However, the cells composing these two groups did present distinct features regarding their size and inner structure (scattering properties) as well as their nucleic acid content (SYBRGreen fluorescence, see Fig. 2). At TS site, the HNA cluster represented 63.7% of the total cell count, belonging to the high (58%) and low (5.7%) side scatter groups (Fig. 2). The LNA cluster represented 32.0% of the total cell count also dispatched between the high (20.8%) and low (11.2%) side scatter groups (Fig. 2). At M1 site, the HNA cluster represented 35.9% of the total cell count, belonging to the high (23.6%) and low (12.3%) side scatter groups (Fig. 2). The LNA cluster represented 63.5% of the total cell count also dispatched between the high (31.0%) and low (32.5%) side scatter groups (Fig. 2). In TS, HNA cells were more abundant than LNA ones. It was the opposite in M1 with also similar differences regarding the structure signal within HNA and LNA. An additional important difference between TS and M1 is the presence of the unidentified group (UG) observed in TS but not in M1, characterized by a larger size signal and a LNA content. To minimize the overlapping of HNA and LNA clusters, sorting was restricted to their core, cells selected for sorting in each core represented about 20% of the total cell count.

Flow cytometry resolution of heterotrophic prokaryotic cell groups derived from typical cytograms of heterotrophic prokaryotes sampled on 3 October 2009 in M1 and TS ponds of the Sfax solar saltern. In the dual-parameter dot plot representing the side scatter (structure) versus forward scatter (size) in arbitrary units (a.u.), the main (>95% of total cell count) cluster was divided in two groups with respect to the structure signal, using the same windows for TS and M1. An additional unidentified group (UG) was singled out in TS but was absent in M1. Each selected group was further plotted individually as green-fluorescence in arbitrary unit (a.u.) of the nucleic acid stain SYBRGreen™ versus forward scatter (size), revealing an additional heterogeneity with respect to the structure signal. The UG group in TS appears as of LNA type but presents larger size and structure signals than the main HNA and LNA groups
Phylogenetic analysis of HNA and LNA cell subgroups from M1 and TS sites
The differences between the TS and M1 ponds in terms of physico-chemical characteristics led to the question as to whether they were also different in terms of species composition and population structure. To this end, we analyzed the biodiversity of LNA and HNA, which were the two most abundant groups (>90% of the total cell counts, Fig. 2) present at the studied sites (M1 and TS). Two libraries were generated, one by the use of 16S rRNA bacterial primers (fD1 and rP2) and one by the use of 16S rRNA universal archaeal primers (21f and 958r).
Although the archaeal 16S rRNA gene was successfully amplified in all samples, we failed to amplify the bacterial 16S rRNA gene in the M1-HNA and TS-LNA samples suggesting that if present, the bacterial population is at very low amount within these samples (Supplementary material, Fig. S1).
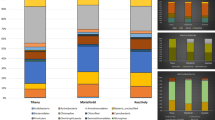
We used the classifier tool version 2.2 of the RDP to perform preliminary taxonomic affiliations of the successfully amplified 40 bacterial and 76 archaeal clones with a confidence threshold of 95% (Table S1; Fig. 3). The use of a lower confidence threshold (down to 85%) gave similar results and did not significantly decrease the proportion of unaffiliated clones (Table S1). The taxonomic affiliations provided by the RDP were verified by phylogenetic analyses (Fig. 4). First, we observed that Bacteria were present only in one cell fraction (HNA fraction, in TS or LNA fraction, in M1) and that Archaea tend to cluster into the two distinct cell fractions. In order to analyze prokaryotic biodiversity in each pond, we decide to pool results from archaeal or bacterial phylogenetic 16 rRNA sequences retrieved from both HNA and LNA fractions. As expected only Archaea affiliated to Halobacteriales order (phylum Euryarchaeota class Halobacteria) were identified in the Archaea population (Figs. 3, 4; Table S1; Antón et al. 2000; Benlloch et al. 2001; Rosselló-Móra et al. 2003; Bolhuis et al. 2004; Walsby 2005; Fendrihan et al. 2006; Maturrano et al. 2006; Baati et al. 2008). However, differences existed between the two ponds. For example, although Halorubrum and Haloquadratum representatives were found in significant proportions (23 and 16%, Figs. 3, 4) in the M1 pond, most archaeal clones (61%) could not be affiliated to cultivated halobacteriales lineages. In contrast, samples from the TS pond were dominated by phylotypes belonging to the Haloquadratum genus (49%, Fig. 3), whereas Halorubrum and unaffiliated clones represented only 21 and 30% of the archaeal population. Compared with Archaea, Bacteria were much less diverse and the difference between the two ponds was more marked. The bacterial population of the M1 pond was composed exclusively by clones affiliated to Phyllobacterium (94%) and Variovorax (6%). This was surprising because these two genera belonging to Alpha- and Beta-proteobacteria divisions are not usually found in halophilic environments (Rojas et al. 2001; Alavi et al. 2001; Willems et al. 1991). In contrast, all bacterial 16S rRNA retrieved from the TS pond represented a single genus, Salinibacter (Bacteroidetes phylum, Sphingobacteria class).
We next aimed to examine the differences between HNA and LNA sub-populations in terms of heterotrophic prokaryotes diversity. As shown in Fig. 3, archaeal sequences corresponding to unclassified Halobacteriales were more abundant in M1-HNA than in M1-LNA, suggesting that HNA population of this pond was mainly composed of an unknown genus. Within HNA Archaea affiliated to the Haloquadratum genus, cells appear to be present in large amounts in HNA in M1 compared with TS (40 vs. 31% of Haloquadratum clones). On the other hand, within Archaea affiliated to Halorubrum genus, cells appear to be much more present in HNA in TS compared with M1 (14 vs. 57% of Halorubrum clones) (see Table S1).

Genus-level diversity based on clone libraries obtained from the most abundant cell groups in M1 and TS ponds. In this study, we used the Naïve Bayesian Classifier for Rapid Assignment of 16S rRNA sequences of HNA and LNA groups, from both TS and M1, into bacterial and archaeal taxonomy according to 95% confidence threshold. All sequences with a confidence threshold of less than 95% were considered as unclassified Bacteria or Archaea and they could be novel representatives of a new genus or species
In the Bacteria analysis (Fig. 3), differences were detected only in LNA for M1 and HNA for TS indicating a clear difference in terms of Bacteria adaptation in these two ponds. Whereas most bacterial sequences from M1 (LNA) (94%) were clustered into Proteobacteria phylum (Alphaproteobacteria class Phyllobacterium genus), all bacterial sequences retrieved from TS (HNA) were clustered into Bacteroidetes phylum (Sphingobacteria class Salinibacter genus).

Bayesian phylogenetic trees of archaeal (a) and bacterial (b) 16S rRNA sequences. Detailed trees are shown as Supplementary material (Figure S2 and Figure S3). Numbers at nodes represent posterior probabilities. For clarity only relevant values are shown. The scale bar represents the average number of substitutions per site
Discussion
In this study, we combined FCM and phylogenetic analyses after cell sorting to study heterotrophic prokaryote groups from crystallizer and non-crystallizer ponds of a coastal solar saltern in Sfax (Tunisia).
Total cell counts per mL (around 107) were evaluated by FCM in the crystallizer pond, TS, or in the non-crystallizer pond, M1, and were similar to those reported for other such extreme salt environments (Guixa-Boixareu et al. 1996; Oren et al. 1996; Antón et al. 1999, 2000; Ochsenreiter et al. 2002; Mutlu et al. 2008). Considering the lack of predation and the high nutrient levels, the high and similar microbial abundance in such diverse crystallizer ponds across the world suggests that salinity is the main factor controlling microbial abundance (Pedrós-Alió et al. 2000).
Archaeal 16S rRNA gene clone libraries indicated that in both M1 and TS ponds, the Haloquadratum and Halorubrum genera are abundant components of the prokaryotic community. This supports the view that Haloquadratum is widespread and often dominates within the microbial community (Benlloch et al. 2001, 2002; Antón et al. 2002; Oren et al. 2006; Ochsenreiter et al. 2002; Mutlu et al. 2008; Baati et al. 2008). Previous studies have also reported Halorubrum to be the most abundant genus in various salterns and salt lakes where Haloquadratum is not dominant or absent (Burns et al. 2004b; Xu et al. 2007; Pasić et al. 2005, 2007). These two major groups of halobacteriales have been recently found to be dominant in three, geographically distant, Australian crystallizer ponds (Oh et al. 2010). These observations agree with the findings that show representatives of the genus Halorubrum and Haloquadratum to be well adapted to extremely high salt concentrations (close or at the saturation point) (Bolhuis et al. 2004; Burns et al. 2004a, 2007; Papke et al. 2007).
Bacterial 16S rRNA gene clone libraries indicated that the genera Phyllobacterium and Salinibacter dominate the Bacteria community in M1 and TS ponds, respectively. Interestingly, unexpected presence of the representatives of Phyllobacterium genus could be explained by the presence of Dinoflagellates, recently observed by Elloumi et al. (2009), in six different salt ponds of the solar saltern of Sfax, among which M2 pond is near the M1 pond of the current study. Indeed, physical association and symbiosis of representatives of Phyllobacterium genus and Dinoflagellate cells have been reported (Alavi et al. 2001).
The presence of representatives of Salinibacter genus in the TS pond was unsurprising because they also comprise between 5 and 30% of the total prokaryotic community in other crystallizer ponds from artificial solar salterns (Elevi Bardavid and Oren 2008; Antón et al. 2000, 2002).
Interestingly, Baati et al. (2008) performed within the same coastal solar saltern in Sfax (Tunisia), 16S rRNA gene sequences analysis in 3 ponds including the crystallizer pond, S5, which appears similar to the TS pond in terms of physico-chemical characteristics and localization. And, our results of the phylogenetic analyses of Bacteria and Archaea from the TS pond agreed with those reported by Baati et al. (2008) for the S5 pond. Indeed, within Archaea, the genera Haloquadratum and Halorubrum were most abundant in both studies, whereas within Bacteria, the genus Salinibacter was most abundant.
The presence of HNA and LNA bacterial groups is a typical FCM phenomenon based on cellular properties observed by FCM analysis after nucleic acid staining. Detection of HNA and LNA Bacteria via FCM has been reported over a wide range of aquatic ecosystems covering large environmental gradients (Troussellier et al. 1999; Gasol et al. 1999; Gasol and del Giorgio 2000; Lebaron et al. 2001; Andreatta et al. 2004; Thyssen et al. 2005; Longnecker et al. 2006; Sherr et al. 2006; Schapira et al. 2010). Here, we show for the first time that this phenomenon could also be extended to archaeal populations.
Ecological or physiological significance of LNA or HNA prokaryotic cells is far from obvious. The relationship between HNA and LNA cell clusters remains elusive and four hypotheses have been proposed (see Bouvier et al. 2007 and references therein). The first proposal is that the HNA fraction represents active and growing microorganisms, whereas those in the LNA fraction are inactive (dormant, injured or dead cells) and originate from the HNA fraction. Another hypothesis is that the LNA fraction encompasses a wide range of physiological states. These include healthy and active cells, and inactive, dormant, and injured cells, all of which share a similar baseline range of nucleic acid, as demonstrated by FCM. In this scenario, HNA cells would simply originate from active LNA cells that are undergoing fast cell division, have replicated their DNA or acquired multiple genome copies. According to these two hypotheses, no differences in taxonomic diversity are expected between the LNA and HNA fractions. In contrast, HNA and LNA have been proposed to represent distinct communities with little or no interaction between them. A final scenario assumes that dynamic exchanges between the two fractions (i.e. passage of HNA to the LNA fraction, and vice versa) exist, and are distinct to each of the fractions. Based on our phylogenetic results after LNA and HNA cell sorting from TS and M1 samples, we observed that Bacteria follow scenario 3 because they were detectable only in one subgroup, LNA in M1 and HNA in TS. By contrast, Archaea cells retrieved from our samples follow scenario 4 where representatives of each genus were detected in all of the samples. These results suggest that different scenarios can co-exist within the same environment.
Finally, if we assume that HNA cells represent the active prokaryote fraction, the exclusive presence of Salinibacter in the HNA group of the crystallizer pond indicates that representatives of the genus Salinibacter are well adapted to high salt concentrations (above saturation point). Indeed, according to its genome and proteome characteristics, Salinibacter exhibits several features that allow it to function at high salt concentrations. These include a potentially versatile metabolism, broad degradative abilities, several ‘halobacterial-like’ characteristics, and adaptation of proteins to function at high salt concentration (Mongodin et al. 2005). These features allow it to compete for space and available nutrients with Halobacteria, sharing the same environment and carrying out all the prokaryotic activities (Gasol et al. 2004), in particular with Haloquadratum (Elevi Bardavid and Oren 2008).
Moreover, we observed that in M1, 14% of the Halorubrum genus and 40% of the Haloquadratum genus were related to the HNA subgroup (Supplementary material, Table S1). In contrast, 57% of the Halorubrum genus and 31% of the Haloquadratum genus were related to the HNA subgroup in TS (Supplementary material, Table S1). Here also, these observations are consistent with the fact that representatives of the genus Halorubrum and Haloquadratum are well adapted to extremely high salt concentration (close or at the saturation point; Bolhuis et al. 2004; Burns et al. 2004b, 2007). Altogether, our observations are in good agreement with the hypothesis that HNA cells represent mainly the active prokaryotic fraction.
In conclusion, combining FCM and phylogenetic analysis after cell sorting is a suitable tool to characterize microbial communities and their physiological behavior. This combination of cytomic and genomic analysis of microorganisms helps to better understand the functioning of their related ecosystems. Phylogenetic analysis alone applied to the bulk community would have been unable to reveal the complexity of the prokaryotic composition as FCM showed it, with the presence of several cell groups in very low abundance (around 1%) and whose identification will require further work.
References
Alavi M, Miller T, Erlandson K, Schneider R, Belas R (2001) Bacterial community associated with Pfiesteria-like dinoflagellate cultures. Environ Microbiol 3:380–396
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Amdouni R (1990) Etude géochimique des saumures libres, des sédiments et des sels dans la saline de Sfax (Tunisie). Thèse de l’Université Paris IV
Andreatta S, Wallinger MM, Piera J, Catalan J, Psenner R, Hofer JS, Sommaruga R (2004) Tools for discrimination and analysis of lake bacterioplankton subgroups measured by flow cytometry in a high-resolution depth profile. Aquat Microb Ecol 36:107–115
Antón J, Liobet-Brossa E, Rodríguez-Valera F, Amann R et al (1999) Fluorescence in situ hybridization analysis of the prokaryotic community inhabiting crystallizer ponds. Environ Microbiol 1:517–523
Antón J, Rosselló-Móra R, Rodriguez-Valera F, Amman R et al (2000) Extremely halophilic Bacteria in crystallizer ponds from solar salterns. Appl Environ Microbiol 66:3052–3057
Antón J, Oren A, Benlloch S, Rodríguez-Valera F, Amann R, Rosselló-Mora R et al (2002) Salinibacter ruber gen nov., sp. nov., a new species of extremely halophilic Bacteria from saltern crystallizer ponds. Int J Syst Evol Microbiol 52:485–491
Ayadi H, Toumi N, Abid O, Medhioub K, Hammami M, Sime-Ngando T, Amblard C, Sargos D (2002) Etude qualitative et quantitative des peuplements phytoplanctoniques et zooplanctoniques dans les bassins de la saline de Sfax, Tunisie. Rev Sci Eau 15:123–135
Baati H, Guermazi S, Amdouni R, Gharsallah N, Sghir A, Ammar E (2008) Prokaryotic diversity of a Tunisian multipond solar saltern. Extremophiles 12:505–518
Benlloch S, Acinas SG, Antón J, López-López A, Luz SP, Rodríguez-Valera F (2001) Archaeal biodiversity in crystallizer ponds from a solar saltern: culture versus PCR. Microb Ecol 41:12–19
Benlloch S, López-López A, Casamayor EO, Øvreås L, Goddard V, Daae FL, Smerdon G, Massana R, Joint I, Thingstad F, Pedrós-Alió C, Rodríguez-Valera F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol 4:349–360
Bidle KA, Amadio W, Oliveira P, Paulish T, Hicks S, Earnest C (2005) A phylogenetic analysis of haloarchaea found in a solar saltern. BIOS 76:89–96
Bolhuis H, Poele EM, Rodriguez-Valera F (2004) Isolation and cultivation of Walsby’s square archaeon. Environ Microbiol 6:1287–1291
Bouvier T, del Giorgio PA, Gasol JM (2007) A comparative study of the cytometric characteristics of high and low nucleic-acid bacterioplankton cells from different aquatic ecosystems. Environ Microbiol 9:2050–2066
Bowman JP, McCammon SA, Rea SM, McMeekin TA (2000) The microbial composition of three limnologically disparate hypersaline Antarctic lakes. FEMS Microbiol Lett 183:81–88
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004a) Cultivation of Walsby’s square haloarchaeon. FEMS Microbiol Lett 238:469–473
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004b) Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarcheal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol 70:5258–5265
Burns DG, Janssen PH, Itoh T, Kamekura M, Li Z, Jensen G, Rodríguez-Valera F, Bolhuis H, Dyall-Smith ML (2007) Haloquadratum walsbyi gen nov., sp. nov., the square haloarchaeon of Walsby, isolated from saltern crystallizers in Australia and Spain. Int J Syst Evol Microbiol 57:387–392
Button DK, Robertson BR (2001) Determination of DNA content of aquatic bacteria by flow cytometry. Appl Environ Microbiol 67:1636–1645
Button DK, Robertson BR, Juttner F (1996) Microflora of a subalpine lake: bacterial populations, size and DNA distribution, and their dependence on phosphate. FEMS Microbiol Ecol 21:87–101
Casamayor EO, Ferrera I, Cristina X, Borrego CM, Gasol JM (2007) Flow cytometric identification and enumeration of photosynthetic sulphur bacteria and potential for ecophysiological studies at the single-cell level. Environ Microbiol 9:1969–1985
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci USA 89:5685–5689
Elevi Bardavid R, Oren A (2008) Sensitivity of Haloquadratum and Salinibacter to antibiotics and other inhibitors: implications for the assessment of the contribution of Archaea and Bacteria to heterotrophic activities in hypersaline environments. FEMS Microbiol Ecol 63:309–315
Elevi Bardavid R, Ionescu D, Oren A, Rainey FA, Hollen BJ, Bagaley DR, Small AM, McKay C (2007) Selective enrichment, isolation and molecular detection of Salinibacter and related extremely halophilic Bacteria from hypersaline environments. Hydrobiologia 576:3–13
Elloumi J, Carrias JF, Ayadi H, Sime-Ngando T, Bouain A (2009) Communities structure of the planktonic halophiles in the solar saltern of Sfax, Tunisia. Estuar Coast Shelf Sci 81:19–26
Fendrihan S, Legat A, Pfaffenhuemer M, Gruber C, Weidler G, Gerbl F, Stan-Lotter H (2006) Extremely halophilic archaea and the issue of long-term microbial survival. Rev Environ Sci Biotechnol 5:203–218
Gasol JM, del Giorgio PA (2000) Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Sci Mar 64:197–224
Gasol JM, Zweifel UL, Peters F, Fuhrman JH, Hagstrom A (1999) Significance of size and nucleic acid content heterogeneity as measured by flow cytometry in natural planktonic bacteria. Appl Environ Microbiol 65:4475–4483
Gasol JM, Casamayor EO, Joint I, Garde K, Gustavson K, Benlloch S, Díez B, Schauer M, Massana R, Pedrós-Alió C (2004) Control of heterotrophic prokaryotic abundance and growth rate in hypersaline planktonic environments. Aquat Microb Ecol 34:193–206
Grégori G, Citterio S, Ghiani A, Labra M, Sgorbati S, Brown S, Denis M (2001) Resolution of viable and membrane-compromised bacteria in freshwater and marine systems based on analytical flow cytometry and nucleic acid double staining. Appl Environ Microbiol 76:4662–4670
Guixa-Boixareu N, Calderon-Paz JI, Heldal M, Bratbak G, Pedrós-Alió C et al (1996) Viral lysis and bacterivory as prokaryotic loss factors along a salinity gradient. Aquat Microb Ecol 11:215–227
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Jellett JF, Li W, Dickie P, Boraie A, Kepkay P (1996) Metabolic activity of bacterioplankton communities assessed by flow cytometry and single carbon substrate utilization. Mar Ecol Prog Ser 136:213–225
Jiang H, Dong H, Zhang G, Yu B, Chapman LR, Fields MW et al (2006) Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl Environ Microbiol 72:3832–3845
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lebaron P, Parthuisot N, Catala P (1998) Comparison of blue nucleic acid dyes for flow cytometric enumeration of bacteria in aquatic systems. Appl Environ Microbiol 64:1725–1730
Lebaron P, Servais P, Agogué H, Courties C, Joux F (2001) Does the nucleic acid content of individual bacteria cells allow us to discriminate between active and inactive cells in aquatic systems. Appl Environ Microbiol 67:1775–1782
Legendre L, Courties C, Troussellier M (2001) Flow cytometry in oceanography 1989–99: environmental challenges and research trends. Cytometry 44:164–172
Longnecker K, Sherr BF, Sherr EB (2005) Activity and phylogenetic diversity of bacterial cells with high and low nucleic acid content and electron transport system activity in an upwelling ecosystem. Appl Environ Microbiol 71:7737–7749
Longnecker K, Sherr BF, Sherr E (2006) Variation in cell-specific rates of leucine and thymidine incorporation by marine bacteria with high and with low nucleic acid content off the Oregon coast. Aquat Microb Ecol 43:113–125
Marie D, Partensky F, Jacquet S, Vaulot D (1997) Enumeration and cell cycle analysis of natural populations of marine picoplankton by flow cytometry using the stain SYBR Green I. Appl Environ Microbiol 63:186–193
Marie D, Simon N, Guillou L, Partensky F, Vaulot D (2000) Flow cytometry analysis of marine picoplankton. In: Diamond RA, DeMaggio S (eds) Living color: protocols in flow cytometry and cell sorting. Springer, Berlin, pp 421–454
Maturrano L, Santos F, Antón J, Rosselló-Móra R et al (2006) Microbial diversity in Maras saltern, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol 72:3887–3895
Mongodin EF, Nelson KE, Daugherty S, Deboy RT, Wister J, Khouri H, Weidman J, Walsh DA, Papke RT, Sanchez Perez G, Sharma AK, Nesbø CL, MacLeod D, Bapteste E, Doolittle WF, Charlebois RL, Legault B, Rodriguez-Valera F (2005) The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc Natl Acad Sci USA 102:18147–18152
Mutlu MB, Martínez-García M, Santos F, Peña A, Guven K, Antón J (2008) Prokaryotic diversity in Tuz Lake, a hypersaline environment in inland Turkey. FEMS Microbiol Ecol 65:474–483
Nishimura Y, Kim C, Nagata T (2005) Vertical and seasonal variations of bacterioplankton subgroups with different nucleic acid contents: possible regulation by phosphorus. Appl Environ Microbiol 71:5828–5836
Ochsenreiter T, Pfeifer F, Schleper C (2002) Diversity of Archaea in hypersaline environments characterized by molecular-phylogenetic and cultivation studies. Extremophiles 6:267–274
Oh D, Porter K, Russ B, Burns D, Dyall-Smith M (2010) Diversity of Haloquadratum and other haloarchaea in three, geographically distant, Australian saltern crystallizer ponds. Extremophiles 14:161–169
Oren A (2002a) Molecular ecology of extremely halophilic Archaea and Bacteria. FEMS Microbiol Ecol 39:1–7
Oren A (2002b) Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol 28:56–63
Oren A, Rodríguez-Valera F (2001) The contribution of Salinibacter species to the red coloration of saltern crystallizer ponds. FEMS Microbiol Ecol 36:123–130
Oren A, Duker S, Ritter S (1996) The polar lipid composition of Walsby’s square bacterium. FEMS Microbiol Lett 138:135–140
Oren A, Pri-El N, Shapiro O, Siboni N (2006) Buoyancy studies in natural communities of square gas-vacuolate archaea in saltern crystallizer ponds. Saline Syst 2:4
Pan LA, Zhang LH, Zhang J, Gasol JM, Chao M (2005) On-board flow cytometry observation of picoplankton community structure in the East China Sea during the fall of different years. FEMS Microbiol Ecol 52:243–253
Papke RT, Zhaxybayeva O, Feil EJ, Sommerfeld K, Muise D, Doolittle WF (2007) Searching for species in haloarchaea. Proc Natl Acad Sci USA 104:14092–14097
Pasić L, Bartual SG, Ulrih NP, Grabnar M, Velikonja BH (2005) Diversity of halophilic archaea in the crystallizers of an Adriatic solar saltern. FEMS Microbiol Ecol 54:491–498
Pasić L, Ulrih NP, Črnigoj M, Grabnar M, Velikonja BH (2007) Haloarchaeal communities in the crystallizers of two Adriatic solar salterns. Can J Microbiol 53:8–18
Pedrós-Alió C, Calderón-Paz JI, Gasol JM (2000) Comparative analysis shows that bacterivory, not viral lysis, controls the abundance of heterotrophic prokaryotic plankton. FEMS Microbiol Ecol 32:157–165
Philippe H (1993) MUST, a computer package of Management Utilities for Sequences and Trees. Nucleic Acids Res 21:5264–5272
Rojas A, Holgwin G, Glick BR, Bashan Z (2001) Synergism between Phyllobacterium sp. (N2-fixer) and Bacillus licheniformis (P-solubilizer), both from a semiarid mangrove rhizosphere. FEMS Microbiol Ecol 35:181
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rosselló-Móra R, Lee N, Antón J, Wagner M et al (2003) Substrate uptake in extremely halophilic microbial communities revealed by microautoradiography and fluorescence in situ hybridization. Extremophiles 7:409–413
Schapira M, Buscot MJ, Pollet T, Leterme SC, Seuront L (2010) Distribution of picophytoplankton communities from brackish to hypersaline waters in a South Australian coastal lagoon. Saline Syst 6:2–15
Servais P, Casamayor EO, Courties C, Catala P, Parthuisot N, Lebaron P (2003) Activity and diversity of bacterial cells with high and low nucleic acid content. Aquat Microb Ecol 33:41–51
Sherr EB, Sherr BF, Longnecker K (2006) Distribution of bacterial abundance and cell-specific nucleic acid content in the Northeast Pacific Ocean. Deep Sea Res Part I Oceanogr Res Pap 53:713–725
Thyssen M, Lefêvre D, Caniaux G, Ras J, Dugrais L, Fernandez Ibanez C, Michel D (2005) Spatial distribution of heterotrophic bacteria in the North East Atlantic (POMME study area) during spring 2001. J Geophys Res 110:CO7S16. doi:10.1029/2004JC002670
Troussellier M, Courties C, Lebaron, Servais P et al (1999) Flow cytometric discrimination of bacterial populations in seawater based on SYTO 13 staining of nucleic acids. FEMS Microbiol Ecol 29:319–330
Walsby AE (2005) Archaea with square cells. Trends Microbiol 13:193–195
Wang Y, Hammes F, Boon N, Chami M, Egli T (2009) Isolation and characterization of low nucleic acid (LNA)-content bacteria. ISME J 3:889–902
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Willems A, De Ley J, Gillis M, Kersters K et al (1991) Comamonadaceae, a new family encompassing the acidovorans rRNA complex, including Variovorax paradoxus gen. nov., comb. nov., for Alcaligenes paradoxus (Davis) 1969. Int J Syst Bacteriol 41:445–450
Xu X, Wu M, Wu Y, Zhang H (2007) Culturable halophilic archaeal diversity of Ayakekumu salt lake located in Xinjiang, China. Acta Ecol Sinica 27:3119–3123
Zubkov MV, Fuchs BM, Burkill PH, Amann R (2001) Comparison of cellular and biomass specific activities of dominant bacterioplankton groups in stratified waters of the Celtic Sea. Appl Environ Microbiol 67:5210–5218
Zubkov MV, Fuchs BM, Tarran GA, Burkill PH, Amann R (2002) Mesoscale distribution of dominant bacterioplankton groups in the northern North Sea in early summer. Aquat Microb Ecol 29:135–144
Acknowledgments
This study was supported by the CMCU committee (project No: 07S0903). We thank the Tunisian COTUSAL salt company in Sfax for permission to access to the saltern and the laboratory of chemical analysis at the ENIS for doing all major cations dosage. C. Brochier-Armanet is supported by an “Action Thématique et Incitative sur Programme (ATIP)” of the French Centre National de la Recherche Scientifique (CNRS).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Oren.
H. Trigui and S. Masmoudi are co-first authors and contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Trigui, H., Masmoudi, S., Brochier-Armanet, C. et al. Characterization of heterotrophic prokaryote subgroups in the Sfax coastal solar salterns by combining flow cytometry cell sorting and phylogenetic analysis. Extremophiles 15, 347–358 (2011). https://doi.org/10.1007/s00792-011-0364-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-011-0364-5