
Abstract
Large DNA viruses in the phylum Nucleocytoviricota, sometimes referred to as “giant viruses” owing to their large genomes and virions, have been the subject of burgeoning interest over the last decade. Here, we describe recently adopted taxonomic updates for giant viruses within the order Imitervirales. The families Allomimiviridae, Mesomimiviridae, and Schizomimiviridae have been created to accommodate the increasing diversity of mimivirus relatives that have sometimes been referred to in the literature as “extended Mimiviridae”. In addition, the subfamilies Aliimimivirinae, Megamimivirinae, and Klosneuvirinae have been established to refer to subgroups of the Mimiviridae. Binomial names have also been adopted for all recognized species in the order. For example, Acanthamoeba polyphaga mimivirus is now classified in the species Mimivirus bradfordmassiliense.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Large dsDNA viruses belonging to the phylum Nucleocytoviricota, often referred to as “giant viruses”, include the largest viruses that have yet been characterized, both in terms of virion size and genome length [1]. Of specific interest are viruses in the order Imitervirales, which are particularly abundant and widespread in ecosystems around the globe [2,3,4,5]. Despite their importance, until recently, only two members of the Imitervirales were classified by the International Committee on Taxonomy of Viruses (ICTV): Acanthamoeba polyphaga mimivirus (APMV) and Cafeteria roenbergensis virus (CroV). Both of these viruses belong to the family Mimiviridae, but a large number of recently sequenced viruses that are only distantly related to either APMV or CroV, sometimes referred to as “extended Mimiviridae” or “extended family Mimiviridae” [6, 7], have recently been reported but not classified. Hence, there has been an urgent need to update the taxonomy of the order Imitervirales to reflect current knowledge on these viruses by classifying those with recently sequenced genomes and organizing them in a hierarchical order by establishing new families, subfamilies, and genera, as needed. Here, we describe taxonomic updates for giant viruses in the order Imitervirales that have recently been adopted by the ICTV.
Phylogenomic analysis
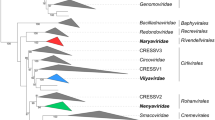
We analyzed 43 genomes of viruses that are, or are related to, current members of the order Imitervirales. We used a concatenated phylogenetic approach (Fig. 1) and also examined pairwise average nucleotide identity (ANI). The concatenated tree is based on seven marker genes: DNA polymerase family B (PolB), RNA polymerase large subunit (RNAPL), A32 packaging enzyme (A32), topoisomerase family II (TopoII), virus late transcription factor 3 (VLTF3), transcription factor IIB (TFIIB), and a superfamily II helicase (SFII). These marker genes have previously been benchmarked and shown to provide high-fidelity phylogenetic trees for viruses belonging to the phylum Nucleocytoviricota [8]. Families, subfamilies, and genera are demarcated such that they have approximately equivalent phylogenetic breadths (i.e., distance from the root). We only describe classification of viral isolates here, but for our concatenated phylogenetic approach, we also included metagenome-derived viral genome sequences. This was done to improve phylogenetic reconstruction by providing additional evolutionary context and to avoid long branches that arise when only isolates are used. For this purpose, a manually selected set of metagenome-derived genome sequences were obtained from the Giant Virus Database (https://faylward.github.io/GVDB/).

Phylogeny of the members of the order Imitervirales, based on an alignment of seven concatenated marker genes (see main text for details). Viruses classified in new or renamed species are shown in bold. Reference genome sequences were obtained from the Giant Virus Database (https://faylward.github.io/GVDB/). The tree was constructed using the ncldv_markersearch workflow (https://github.com/faylward/ncldv_markersearch), which has been described previously [8]
Guidelines used for binomial species names
Newly adopted family names have the suffix “-mimiviridae” to denote evolutionary relatedness to the Mimiviridae (Allomimiviridae, Mesomimiviridae, Schizomimiviridae) and to acknowledge the original name of this group of viruses proposed in 2008 [9]. New subfamilies are either named by officially adopting names that are commonly used in the literature (Megamimivirinae, Klosneuvirinae) or using a simple Latin prefix (Aliimimivirinae; alii- Latin for “other”). Subfamilies were created only for the family Mimiviridae because the most viral isolates are available for this family. For species, we adopted binomial names. Genus names refer to the names of Titans or gods in Greek mythology (Biavirus, Kratosvirus, Heliosvirus, Oceanusvirus, Rheavirus, Tethysvirus, and Theiavirus) unless another genus name was already in common usage in the literature (Megavirus, Moumouvirus, Fadolivirus, Yasminevirus, Cotonvirus, and Tupanvirus). The second name in the binomial refers to features or geography where viruses were sampled or isolated (i.e., sinusmexicani denoting isolation from the Gulf of Mexico). For consistent use of binomial names, we also renamed the currently recognized species Acanthamoeba polyphaga mimivirus and Cafeteria roenbergensis virus to Mimivirus bradfordmassiliense and Rheavirus sinusmexicani, respectively, to fit the new Linnaean binomial format. A full list of all official binomial names can be found in Table 1.
Proposed families
Family Allomimiviridae
“Allo-“ from Greek allos “other, different”. This family contains the species Heliosvirus raunefjordenense and Oceanusvirus kaneohense. Members of both species are marine viruses that infect green algae (Pyramimonas orientalis and Tetraselmis sp., respectively). The name denotes the broad phylogenetic relationship of this family to the family Mimiviridae.
Family Schizomimiviridae
“Schizo-” from Greek schizo, “split”. This family contains Biavirus raunefjordenense and Kratosvirus quantuckense to classify marine viruses that infect haptophyte and heterokont hosts, respectively. The name refers to the splitting of these viruses from others that have commonly been referred to “extended Mimiviridae”.
Family Mesomimiviridae
“Meso”- from Greek mesos, “middle”. This family contains Tethysvirus hollandense, T. ontarioense, and T. raunefjordenense, all of which include aquatic viruses that infect haptophytes. These viruses form a highly supported monophyletic clade in our concatenated phylogeny, and we therefore propose that they should be classified in the same genus. The name refers to the previous description of these viruses as “extended Mimiviridae” and reflects their medium-sized genome lengths, as compared to other Imitervirales members. This lineage was previously suggested to represent a subfamily within the family Mimiviridae with the name Mesomimivirinae [6], but analysis of the concatenated phylogeny demonstrates that this group can be clearly distinguished from the Mimiviridae and that a new, family-level rank is appropriate (Fig. 1).
Proposed subfamilies
Three new subfamilies have been created within the family Mimiviridae, all of which form highly supported clades of approximately the same phylogenetic breadth. The literature often refers to the subfamilies Megamimivirinae and Klosneuvirinae, and we therefore felt it is appropriate to formalize these names and also to introduce the subfamily Aliimimivirinae such that consistent reference can be made to all subclades within this family.
Subfamily Megamimivirinae
This subfamily includes members of the genera Tupanvirus, Cotonvirus, Mimivirus, Megavirus, and Moumouvirus, all of which infect amoebae. The name is already used frequently in the literature (e.g., [6]) and reflects their genome lengths in the megabase-pair range.
Subfamily Klosneuvirinae
This subfamily includes the viruses Fadolivirus FV1/VV64 (species Fadolivirus algeromassiliense), Yasminevirus sp. GU-2018 (species Yasminevirus saudimassiliense), and Bodo saltans virus (species Theiavirus salishense). The first two of these infect amoeba hosts, while the third infects a kinetoplastid phagotrophic protozoan (Bodo saltans). The name is already used frequently in the literature (e.g., [10]).
Subfamily Aliimimivirinae
“Alii-”, Latin for “other”. This subfamily contains a single species, Rheavirus sinusmexicani, to classify Cafeteria roenbergensis virus (CroV), which infects a flagellate protozoan (Cafeteria roenbergensis).
Demarcation criteria
Members of the same species have pairwise ANI > 95% for >75% of the predicted genes in each genome. The species Mimivirus bradfordmassiliense, Moumouvirus ??moumou, Megavirus chilense, and Tethysvirus hollandense include multiple viral isolates that fit these criteria (Table 1).
Families and genera are defined as monophyletic clades in the concatenated tree that have high bootstrap support (>80%) and approximately equivalent phylogenetic breadth (Fig. 1). Our family-level demarcations are consistent with a phylogenomic framework that has been proposed recently [8].
This phylogenetic methodology should be adequate for adding new taxonomic levels in the future. Although it relies on seven marker genes (PolB, A32, SFII, VLTF3, RNAPS, TopoII, and TFIIB), genome sequences can be included even if they encode only a subset of these genes. For example, prasinoviruses that lack RNA polymerase subunits were still included using this phylogenetic approach, and they had robust phylogenetic placement [8]. Therefore, novel viral lineages that are discovered in the future could still be included using these methods even if their genomic composition differed slightly from the viruses examined here. We propose that new families should be created once the first complete genome sequence is available for a viral lineage that represents a phylogenetic breadth consistent with the existing families. Most of these complete genome sequences will likely be derived from isolated viruses, but in the future it may be possible for high-quality genome sequences that are suitable for classification to be obtained using culture-independent methods, which is in full agreement with recently adopted ICTV standards and recommendations for development of a universal sequence-based virus taxonomy [11, 12].
Conclusions
Recent studies have shown that members of the family Mimiviridae and other viruses in the order Imitervirales are abundant in a wide range of ecosystems across the planet [2, 4, 5, 13, 14]. These viruses infect diverse protists from across the eukaryotic tree of life, including amoebae, green algae, euglenoids, stramenopiles, and haptophytes [15,16,17,18,19]. Furthermore, members of the order Imitervirales typically have large genomes that encode proteins with a wide range of complex functions, including rhodopsins, cytoskeletal structural proteins, and predicted components of the TCA cycle and glycolysis [4, 20,21,22,23,24]. As the number of known viruses within the order Imitervirales has grown, it has become clear that the establishment of several new families is needed to classify the burgeoning diversity within this group. For example, a recent study using primarily metagenome-derived giant virus genome sequences suggested that the order Imitervirales contains at least 11 families [8]. The newly adopted families Allomimiviridae, Mesomimiviridae, and Schizomimiviridae are therefore likely to be the first of several new families that will be demarcated within the order Imitervirales as new viruses are isolated and complete genome sequences become available.
References
Abergel C, Legendre M, Claverie J-M (2015) The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol Rev 39:779–796
Endo H, Blanc-Mathieu R, Li Y et al (2020) Biogeography of marine giant viruses reveals their interplay with eukaryotes and ecological functions. Nat Ecol Evol 4:1639–1649
Monier A, Larsen JB, Sandaa R-A et al (2008) Marine mimivirus relatives are probably large algal viruses. Virol J 5:12
Moniruzzaman M, Martinez-Gutierrez CA, Weinheimer AR, Aylward FO (2020) Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat Commun 11:1710
Schulz F, Roux S, Paez-Espino D et al (2020) Giant virus diversity and host interactions through global metagenomics. Nature 578:432–436
Gallot-Lavallée L, Blanc G, Claverie J-M (2017) Comparative genomics of Chrysochromulina ericina virus and other microalga-infecting large DNA viruses highlights their intricate evolutionary relationship with the established Mimiviridae family. J Virol. https://doi.org/10.1128/JVI.00230-17
Koonin EV, Yutin N (2018) Multiple evolutionary origins of giant viruses. F1000Res. https://doi.org/10.12688/f1000research.16248.1
Aylward FO, Moniruzzaman M, Ha AD, Koonin EV (2021) A phylogenomic framework for charting the diversity and evolution of giant viruses. PLoS Biol 19:e3001430
La Scola B, de Lamballierie X, J-M C, et al (2005) Create new family Mimiviridae. Taxonomic proposal. https://ictv.global/ictv/proposals/2005.004F.04.Mimiviridae.pdf
Andreani J, Schulz F, Di Pinto F et al (2021) Morphological and genomic features of the new Klosneuvirinae isolate Fadolivirus IHUMI-VV54. Front Microbiol. https://doi.org/10.3389/fmicb.2021.719703
Simmonds P, Adams MJ, Benkő M et al (2017) Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol 15:161–168
Simmonds P, Adriaenssens EM, Zerbini FM et al (2023) Four principles to establish a universal virus taxonomy. PLoS Biol 21:e3001922
Ha AD, Moniruzzaman M, Aylward FO (2023) Assessing the biogeography of marine giant viruses in four oceanic transects. ISME Commun 3:43
Schulz F, Alteio L, Goudeau D et al (2018) Hidden diversity of soil giant viruses. Nat Commun 9:4881
Fischer MG, Allen MJ, Wilson WH, Suttle CA (2010) Giant virus with a remarkable complement of genes infects marine zooplankton. Proc Natl Acad Sci U S A 107:19508–19513
Deeg CM, Chow C-ET, Suttle CA (2018) The kinetoplastid-infecting Bodo saltans virus (BsV), a window into the most abundant giant viruses in the sea. Elife. https://doi.org/10.7554/eLife.33014
Stough JMA, Yutin N, Chaban YV et al (2019) Genome and environmental activity of a virus and its virophages. Front Microbiol 10:703
Schvarcz CR, Steward GF (2018) A giant virus infecting green algae encodes key fermentation genes. Virology 518:423–433
Blanc-Mathieu R, Dahle H, Hofgaard A et al (2021) A persistent giant algal virus, with a unique morphology, encodes an unprecedented number of genes involved in energy metabolism. J Virol. https://doi.org/10.1128/JVI.02446-20
Ha AD, Moniruzzaman M, Aylward FO (2021) High transcriptional activity and diverse functional repertoires of hundreds of giant viruses in a coastal marine system. mSystems 6:e0029321
Da Cunha V, Gaia M, Ogata H et al (2022) Giant viruses encode actin-related proteins. Mol Biol Evol. https://doi.org/10.1093/molbev/msac022
Kijima S, Delmont TO, Miyazaki U et al (2021) Discovery of viral myosin genes with complex evolutionary history within plankton. Front Microbiol 12:683294
Yutin N, Koonin EV (2012) Proteorhodopsin genes in giant viruses. Biol Direct 7:34
Moniruzzaman M, Erazo Garcia MP, Farzad R, Ha AD, Jivaji A, Karki S, Sheyn U, Stanton J, Minch B, Stephens D, Hancks DC, Rodrigues RAL, Abrahao JS, Vardi A, Aylward FO (2023) Virologs, viral mimicry, and virocell metabolism: the expanding scale of cellular functions encoded in the complex genomes of giant viruses. FEMS microbiology reviews, 47(5), fuad053. https://doi.org/10.1093/femsre/fuad053
Acknowledgements
We are grateful to Bernard La Scola, Ruth-Ann Sandaa, Steven Short, Grieg Steward, and Steven Wilhelm for help with clarifying the provenance of various viruses used in this classification.
Funding
FOA was funded by NIH NIGMS award 1R35GM147290-01 and NSF award 2141862.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have not disclosed any competing interests.
Additional information
Handling Editor: Sead Sabanadzovic.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aylward, F.O., Abrahão, J.S., Brussaard, C.P. et al. Taxonomic update for giant viruses in the order Imitervirales (phylum Nucleocytoviricota). Arch Virol 168, 283 (2023). https://doi.org/10.1007/s00705-023-05906-3
Published:
DOI: https://doi.org/10.1007/s00705-023-05906-3