
Abstract
Geminiviruses are plant-infecting, circular single-stranded DNA viruses that have a geminate virion morphology. These viruses infect both cultivated and non-cultivated monocotyledonous and dicotyledonous plants and have a wide geographical distribution. Nine genera had been established within the family Geminiviridae (Becurtovirus, Begomovirus, Capulavirus, Curtovirus, Eragrovirus, Grablovirus, Mastrevirus, Topocuvirus, and Turncurtovirus) as of 2020. In the last decade, metagenomics approaches have facilitated the discovery and identification of many novel viruses, among them numerous highly divergent geminiviruses. Here, we report the establishment of five new genera in the family Geminiviridae (Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus) to formally classify twelve new, divergent geminiviruses.
Similar content being viewed by others

Introduction
Over the past 50 years, plant-infecting geminiviruses (family Geminiviridae) have been amongst the most important pathogens of cultivated plant species in most tropical and subtropical regions of the world [28]. Geminiviruses infect both monocotyledonous and dicotyledonous plants, in some cases without visible symptoms and in others causing symptoms that include different degrees of foliar crinkling, curling, yellowing, distortion, stunting, mosaic and/or striations [28]. The family Geminiviridae includes viruses with circular single-stranded DNA (ssDNA) genomes that are individually encapsidated into 22 × 38 nm twinned quasi-icosahedral virions made up of 110 capsid protein subunits organized as 22 pentameric capsomers [2, 16, 17, 41]. The genomes of geminiviruses can be monopartite, with a single DNA molecule of ~ 2,600–3,600 nucleotides (nt), or bipartite, with two DNA molecules of ~ 2,600 nt (termed DNA-A and DNA-B) for a total genome size of ~ 5,200 nt [38].
As of 2020, there were nine genera (Becurtovirus, Begomovirus, Capulavirus, Curtovirus, Eragrovirus, Grablovirus, Mastrevirus, Topocuvirus, and Turncurtovirus) [36, 38] and two unassigned species, Citrus chlorotic dwarf associated virus [19] and Mulberry mosaic dwarf associated virus [20, 22] in the family Geminiviridae. A framework for species classification based on pairwise genome sequence identity between member viruses, coupled with phylogenetic support, has been established for becurtoviruses [35], begomoviruses [3], capulaviruses [36], curtoviruses [34], eragroviruses [35], grabloviruses [36], mastreviruses [24], topocuviruses [26], and turncurtoviruses [35].
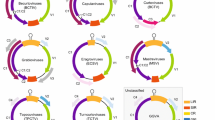
Over the last several years, a number of new geminiviruses have been identified that do not fit the previously established nine genera. These include geminiviruses identified infecting apple trees (Malus domestica Borkh.) [18], camellia (Camellia japonica L.) [40], grapevine (Vitis vinifera L. [1], sea rush (Juncus maritimus Lam.) [5], passion fruit (Passiflora edulis Sims.) [8], tomato (Solanum lycopersicum L.) [7, 33], cleome (Cleome sp.) [7], cactus plants (subfamilies Cactoideae and Opuntioideae in the Cactaceae) [9], white mulberry (Morus alba L.) [20, 22], and paper mulberry (Broussonetia papyrifera (L.) L'Hér. ex Vent.) [27]. Based on the inferred genome organizations of these viruses (Fig. 1) coupled with phylogenetic analysis (Fig. 2), these diverse geminiviruses are now assigned to five new genera: Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus (Figs. 1 and 2, Table 1). Within these genera, twelve species have been established, and species demarcation criteria have been defined.

Illustration of the genome organisation of various geminiviruses. LIR, long intergenic region; SIR, short intergenic region; CR, common region; cp, capsid protein; mp, movement protein; nsp, nuclear shuttle protein; reg, regulatory gene; ren, replication enhancer; rep, replication-associated protein; sd, symptom determinant; ss, silencing suppressor; trap, transactivator protein. The genomes of representative members of the five new genera are indicated by asterisks.

Unrooted neighbor-joining tree inferred from aligned complete genome sequences of representative isolates from the various geminivirus genera. The five new genera are indicated by asterisks. Branches with less than 60% bootstrap support have been collapsed with TreeGraph2 [32].
Genus Citlodavirus
A group of closely related viruses isolated from citrus in China, Thailand, and Turkey [19, 37, 42], Camellia japonica and Camellia sinensis in China [40], Passiflora edulis in Brazil [8], and Broussonetia papyrifera in China [27] has been assigned to the new genus Citlodavirus (Table 1). The genus name Citlodavirus is derived from the “type member” of the genus: citrus chlorotic dwarf associated virus [19]. The genome size of these viruses is approximately 20% larger than that of other known monopartite geminiviruses and ranges from 3639 to 3763 nt (Fig. 1). This greater length of their genomes is due to their predicted mp gene (891–921 nt in size), which likely encodes a protein product homologous to that encoded by the DNA-B component of bipartite begomoviruses (genus Begomovirus) [8].
These viruses have the virion-strand origin of replication nonanucleotide motif ‘TAATATTAC’ and a unique genome arrangement. The virion-sense strand potentially encodes a capsid protein (CP), a movement protein (MP), and two other small hypothetical proteins, referred to as V2 and V3 (Fig. 1). The complementary strand potentially encodes a RepA protein and expresses a replication initiator protein (Rep) from an alternatively spliced complementary-sense transcript (Fig. 1). While the natural vector has not been formally identified yet, it has been proposed that the vector of citrus chlorotic dwarf associated virus (CCDaV) could be a whitefly (Parabemisia myricae) that thrives on woody plants [11, 19]. Phylogenetic analysis of the predicted CP amino acid sequence (Fig. 3) and pairwise sequence comparisons (Fig. 4) show that the citlodavirus CPs are most similar to those of begomoviruses that are transmitted by Bemisia tabaci. The citlodavirus capsid morphology is currently unknown, and further studies are needed to ascertain whether the > 3200-nt-long genomes of these viruses are encapsidated within geminate particles, or whether the particles form alternative structures [30].

Maximum-likelihood phylogenetic trees inferred using PhyML 3 [13], based on the Rep and CP amino acid sequences of representative members of the various genera in the family Geminiviridae. rtREV+G+I (Rep) and rtREV+G+F+I (CP) were used as best-fit substitution models as determined using ProtTest 3 [6]. Branches with < 60% bootstrap support have been collapsed with TreeGraph2 [32].

Pairwise identity matrix of Rep and CP amino acid sequences of representative members of the genera in the family Geminiviridae and four unassigned species determined using SDT v1.2 [25].
An analysis of the distribution of pairwise identities (one minus Hamming distances of pairwise aligned sequences with pairwise deletion of gaps) of known citlodavirus genome sequences (n = 20; Table 1) using SDT v1.2 [25] indicates that a pairwise-identity-based species demarcation threshold that would minimize conflicts (i.e., possible assignments of individual isolates to two or more species) could be placed in the 66-to-90% interval (Fig. 5, Supplementary Data 1). To align the citlodavirus species demarcation threshold with that of the majority of genera of the family Geminiviridae, we have opted to tentatively use 78% as the pairwise identity species demarcation threshold. Citlodaviruses whose genome sequences had less than 78% pairwise identity, coupled with phylogenetic support, would be considered members of new species. Based on these demarcation criteria, four citlodavirus species were established to accommodate the 20 citlodavirus isolates (Table 1). These species are Citrus chlorotic dwarf associated virus (n = 15), Camellia chlorotic dwarf-associated virus (n = 2), Paper mulberry leaf curl virus 2 (n = 2), and Passion fruit chlorotic mottle virus (n = 1). Viruses assigned to a citlodavirus species share between 61% and 66% genome-wide nucleotide sequence identity with members of other species within the genus (Fig. 2).

Distribution of pairwise identity values for the genomes of viruses in the genera Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus, determined using SDT v1.2 [25]. The cyan line represents the species demarcation threshold for each genus.
Regardless of whether the full-genome nucleotide sequence, the inferred Rep amino acid sequence, or the inferred CP amino acid sequence is considered, all viruses belonging to the proposed citlodavirus species group, with 60-100% bootstrap support, with other proposed citlodaviruses (Figs. 2 and 3). The Reps and CPs of the viruses assigned to the genus Citlodavirus share 32-44% and 16-40% amino acid sequence identity, respectively, with those of other geminiviruses (Fig. 4).
Genus Maldovirus
A group of closely related viruses isolated from two dicotyledonous plants, apple tree in China [18] and grapevine in China, Israel, Japan, Hungary, and South Korea [1], and one monocotyledonous plant, sea rush in France [5], has been assigned to the new genus Maldovirus (Table 1). The genus name Maldovirus (Malus domestica virus) is derived from the scientific name of the host plant (Malus domestica Borkh.) of the “type member” of the genus, apple geminivirus 1. All 15 known maldovirus isolates have the same ‘TAATATTAC’ virion-strand origin of replication nonanucleotide sequence motif and share a similar arrangement of five to six open reading frames (ORFs) within their genomes (Fig. 1). While the virion-sense strand potentially encodes a CP and one other small hypothetical protein, referred to as V2 (Fig. 1), the complementary strand potentially encodes a Rep and two or three other small hypothetical proteins, referred to as C2, C3, and C4 (Fig. 1). It is noteworthy that a naturally occurring 1559-nt subgenomic/defective form of grapevine geminivirus A has been isolated from various grapevines housed in germplasm collections in the USA [1]. Grapevine geminivirus A and its defective molecule were both shown to be graft-transmissible [1]. The natural vector of maldoviruses has not been identified to date.
An analysis of the distribution of pairwise identity values for known maldovirus genomes (n = 15; Table 1) indicates that a pairwise-identity-based species demarcation threshold could be placed in the 67-to-97% interval (Fig. 5, Supplementary Data 1). To align the maldovirus species demarcation threshold with that of the majority of genera of the family Geminiviridae, a 78% pairwise identity species demarcation threshold has tentatively been adopted. Therefore, groups of maldoviruses with less than 78% pairwise identity to other maldoviruses but >78% identity to one another, coupled with phylogenetic support for their branching within a separate clade, would be considered members of the same new species.
The 15 maldoviruses can therefore be assigned to three species (Table 1) i.e., Apple geminivirus 1 (n = 1), Grapevine geminivirus A (n = 13), and Juncus maritimus geminivirus 1 (n = 1). Genome sequences within each species share between 62% and 67% genome-wide identity with members of other maldovirus species (Fig. 2). Furthermore, regardless of whether the full-genome nucleotide sequence, the inferred Rep amino acid sequence, or the inferred CP amino acid sequence is considered, all of the viruses belonging to the proposed maldovirus species group, with 88-100% bootstrap support, with other proposed maldoviruses (Figs. 2 and 3). The Reps and CPs of the viruses assigned to the genus Maldovirus share 28-69% and 14-29% amino acid sequence identity, respectively, with those of other geminiviruses (Fig. 4).
Genus Mulcrilevirus
A group of closely related viruses isolated from white mulberry and paper mulberry in China [20, 22, 27] has been assigned to the new genus Mulcrilevirus (Table 1). Specifically, eight mulberry mosaic dwarf associated virus (MMDaV) isolates and one mulberry crinkle leaf virus (MCLV) isolate (Table 1) have been characterized from diseased white mulberry plants displaying crinkle leaf, mosaic and/or dwarfing symptoms [20, 22]. The genus name Mulcrilevirus is derived from the “type member” of the genus: mulberry crinkle leaf virus. All 11 known mulcrilevirus isolates have the same ‘TAATATTAC’ virion-strand origin of replication nonanucleotide sequence motif and an arrangement of ORFs similar to those described previously for other geminiviruses (Fig. 1). The virion-sense strand encodes a putative MP, a CP, and two additional small hypothetical proteins, referred to as V2 and V4 (Fig. 1). The complementary strand of the genome potentially encodes a RepA protein and expresses a Rep protein from a spliced complementary-strand transcript (Fig. 1). It has been proposed recently that MCLV could be transmitted by the leafhopper Tautoneura mori Matsumura [21].
An analysis of the distribution of pairwise identity values for known mulcrilevirus genomes (n = 11; Table 1) indicates that a pairwise-identity-based species demarcation threshold could be placed in the 61-to-97% interval (Fig. 5, Supplementary Data 1). To align the mulcrilevirus species demarcation threshold with that of the majority of genera of the family Geminiviridae, we have tentatively adopted a 78% pairwise identity species demarcation threshold. The MCLV and MMDaV isolates share > 96.9% genome-wide pairwise identity with each other, indicating that they belong to the same species (Fig. 2). Based on these criteria, two species in the genus Mulcrilevirus were established: Mulberry crinkle leaf virus (including isolates of MMDaVs and MCLVs) and Paper mulberry leaf curl virus 1. Isolates of a given Mulcrilevirus species share between 60.5% and 60.8% genome-wide sequence identity with each other (Fig. 2) and cluster together in phylogenetic trees with 63-91% bootstrap support, regardless of whether the full-genome nucleotide sequence, the inferred Rep amino acid sequence, or the inferred CP amino acid sequence is used for the analysis (Figs. 2 and 3). The Reps and CPs of the viruses assigned to the genus Mulcrilevirus share 32-53% and 17-40% amino acid sequence identity, respectively, with those of other geminiviruses (Fig. 4).
Genus Opunvirus
A group of closely related viruses has been identified in asymptomatic New World Cactaceae plants [9] and has been assigned to the new genus Opunvirus (Table 1). Specifically, 79 opuntia virus 1 (OpV1) genome sequences have been determined from cactus plants belonging to 20 different cactus species from both the Cactoideae and Opuntioideae subfamilies and from nine cactus-feeding cochineal insects (Dactylopius sp.) sampled in the USA and Mexico [9] (Table 1). The genus name Opunvirus is derived from the “type member” of the genus: Opuntia virus 1. All 79 known opunvirus genomes have the same ‘TAATATTAC’ virion-strand origin of replication nonanucleotide sequence motif and an arrangement of the six ORFs similar to those described previously for other geminiviruses (Fig. 1). The genome organization of opunviruses resembles that of monopartite begomoviruses. On the complementary strand, the opunvirus sequences encode a Rep, a putative replication enhancer protein (Ren), a putative transactivation protein (TrAP), and a putative symptom determinant protein (C4) (Fig. 1). A CP and a possible MP are encoded on the virion strand. Interestingly, OpV1 sequences were isolated directly from cochineal insects that were associated with the cactus plants from which OpV1 sequences were isolated [9]. However, the transmission of OpV1 has not been unequivocally demonstrated, and controlled insect transmission experiments will be needed to determine if cochineal insects are vectors for opunviruses.
An analysis of the distribution of pairwise identity values for of known OpV1 sequences (n = 79; Table 1) indicates that their genomes share > 78.4% genome-wide pairwise identity with each other (Fig. 5, Supplementary Data 1), and thus, for the moment, they have been assigned to a single species, Opuntia virus 1. We suggest that a 78% species demarcation threshold should also be used for this genus. In addition, OpV1 genome sequences share less than 64.9% identity with all other known geminiviruses within currently established species (Fig. 2).
Regardless of whether the full-genome nucleotide sequence, the inferred Rep amino acid sequence, or the inferred CP amino acid sequence is used for the analysis, all OpV1 isolates cluster together in phylogenetic trees with 100% bootstrap support (Figs. 2 and 3). The Reps and CPs of the viruses assigned to the genus Opunvirus share 30-68% and 15-29% amino acid sequence identity, respectively, with those of other geminiviruses (Fig. 4).
Genus Topilevirus
From tomato and cleome plants sampled in Argentina and Brazil [7, 33], a group of closely related viruses has been identified that has been assigned to the new genus Topilevirus (Table 1). The genus name Topilevirus was derived from the “type member” of the genus: tomato apical leaf curl virus. The genome of topileviruses has a ‘TAATATTAC’ virion-strand origin of replication nonanucleotide motif. A CP, a possible MP, and a protein that possibly regulates relative ssDNA and dsDNA levels (Reg) are encoded on the virion strand (Fig. 1). The complementary strand of the genome potentially encodes a RepA protein, one small hypothetical protein, referred to as C3, and a Rep protein from a spliced complementary-strand transcript (Fig. 1).
Similar to viruses in the genera Becurtovirus, Capulavirus, Citlodavirus, Eragrovirus, Grablovirus, and Mastrevirus, topileviruses have two intergenic regions (large intergenic region [LIR] and small intergenic region [SIR]; Fig. 1). The natural vector of topileviruses is not known. However, using in silico prediction based on capsid protein sequences, it has been proposed that treehoppers in the family Membracidae could be the vector of this virus [33]. Nonetheless, this has not been experimentally confirmed yet.
An analysis of the distribution of pairwise identity values for known topilevirus genomes (n = 5; Table 1) indicates that a pairwise-identity-based species demarcation threshold could be placed in the 65-to-99% interval (Fig. 5, Supplementary Data 1). A 78% pairwise identity species demarcation threshold has tentatively been adopted for topileviruses, and thus the five viruses are assigned to two species, i.e., Tomato apical leaf curl virus (n = 3) and Tomato geminivirus 1 (n = 2) (Table 1). Based on pairwise identity comparisons, all isolates (tomato apical leaf curl virus and tomato geminivirus 1) within each of these species share between 64.2% and 64.9% genome-wide sequence identity with all isolates that have been assigned to other proposed topilevirus species (Fig. 2).
Regardless of whether the full-genome nucleotide sequence, the inferred Rep amino acid sequence, or the inferred CP amino acid sequence is used for the analysis, all topilevirus isolates cluster together in phylogenetic trees with 75-99% bootstrap support (Figs. 2 and 3). The Reps and CPs of the viruses assigned to the genus Topilevirus share 32-70% and 17-35% amino acid sequence identity, respectively, with those of other geminiviruses (Fig. 4).
Concluding remarks
With this report, a total of 10 new geminivirus genera have been established in the last decade to classify diverse members of the family Geminiviridae. These genera have been created to accommodate sequences that have predominantly been identified as a result of improved molecular techniques, including rolling-circle amplification [14] and high-throughput sequencing approaches [23, 29]. Furthermore, a recent study has identified previously unappreciated coding regions within the genome of what is arguably one of the best-studied geminiviruses, the begomovirus tomato yellow leaf curl virus. These coding regions encode small proteins that appear to play key roles in the subcellular localization of viral components and in virulence [12]. What this study indicates is that the genome organizations of all other geminiviruses, including those described here, could also be considerably more complex than can be determined by a cursory accounting of conserved ORFs.
At the time of writing, there remain four geminivirus species that have not been assigned to any established genera: Common bean curly stunt virus, Limeum africanum associated virus, Parsley yellow leaf curl virus, and Polygala garcinii associated virus [5, 15, 39]. These four species are either singletons or do not group with other known geminiviruses (Figs. 2 and 3). Further, we note that there have also been two recent reports of novel geminiviruses that have been identified in olive trees [4] and cacti [10].
Finally, we would like to point out that virus taxonomy is dynamic and not static. As we survey ever-increasing swathes of the geminivirus sequence space, representatives of novel species will be discovered that will demand that we change some of the criteria that we use to classify these viruses. We would like to note that although species names may change within this dynamic taxonomy, the virus names do not change. For example, bean golden mosaic virus will always be bean golden mosaic virus, even though the species name may change. We also would like to remind the reader that a standardized binomial species nomenclature, consisting of the genus name and a free-form species epithet, has been ratified recently by the International Committee on Taxonomy of Viruses (ICTV) [31]. A summary of the current species in the family Geminiviridae is provided in Supplementary Data 2. Thus, we encourage the community to engage with the ICTV Geminiviridae and Tolecusatellitidae Study Group to help smooth the transition from the more free-form virus nomenclature of the past to its new standardised version.
References
Al Rwahnih M, Alabi OJ, Westrick NM, Golino D, Rowhani A (2017) Description of a novel monopartite geminivirus and its defective subviral genome in grapevine. Phytopathology 107:240–251
Bottcher B, Unseld S, Ceulemans H, Russell RB, Jeske H (2004) Geminate structures of African cassava mosaic virus. J Virol 78:6758–6765
Brown JK, Zerbini FM, Navas-Castillo J, Moriones E, Ramos-Sobrinho R, Silva JC, Fiallo-Olive E, Briddon RW, Hernandez-Zepeda C, Idris A, Malathi VG, Martin DP, Rivera-Bustamante R, Ueda S, Varsani A (2015) Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch Virol 160:1593–1619
Chiumenti M, Greco C, De Stradis A, Loconsole G, Cavalieri V, Altamura G, Zicca S, Saldarelli P, Saponari M (2021) Olea Europaea geminivirus: a novel bipartite geminivirid infecting olive trees. Viruses 13:481
Claverie S, Bernardo P, Kraberger S, Hartnady P, Lefeuvre P, Lett JM, Galzi S, Filloux D, Harkins GW, Varsani A, Martin DP, Roumagnac P (2018) From spatial metagenomics to molecular characterization of plant viruses: a geminivirus case study. Adv Virus Res 101:55–83
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Fontenele RS, Lamas NS, Lacorte C, Lacerda ALM, Varsani A, Ribeiro SG (2017) A novel geminivirus identified in tomato and cleome plants sampled in Brazil. Virus Res 240:175–179
Fontenele RS, Abreu RA, Lamas NS, Alves-Freitas DMT, Vidal AH, Poppiel RR, Melo FL, Lacorte C, Martin DP, Campos MA, Varsani A, Ribeiro SG (2018) Passion fruit chlorotic mottle virus: molecular characterization of a new divergent geminivirus in Brazil. Viruses 10:169
Fontenele RS, Salywon AM, Majure LC, Cobb IN, Bhaskara A, Avalos-Calleros JA, Arguello-Astorga GR, Schmidlin K, Khalifeh A, Smith K, Schreck J, Lund MC, Kohler M, Wojciechowski MF, Hodgson WC, Puente-Martinez R, Van Doorslaer K, Kumari S, Verniere C, Filloux D, Roumagnac P, Lefeuvre P, Ribeiro SG, Kraberger S, Martin DP, Varsani A (2020) A novel divergent geminivirus identified in asymptomatic new world Cactaceae plants. Viruses 12:398
Fontenele RS, Salywon AM, Majure LC, Cobb IN, Bhaskara A, Avalos-Calleros JA, Arguello-Astorga GR, Schmidlin K, Khalifeh A, Smith K, Schreck J, Lund MC, Kohler M, Wojciechowski MF, Hodgson WC, Puente-Martinez R, Van Doorslaer K, Kumari S, Oyeniran KA, Verniere C, Filloux D, Roumagnac P, Lefeuvre P, Ribeiro SG, Kraberger S, Martin DP, Varsani A (2021) New world Cactaceae plants harbor diverse geminiviruses. Viruses 13:694
Garnsey SM (1996) Citrus chlorotic dwarf, a new whitefly-transmitted disease in the Eastern Mediterranean region of Turkey. In: International organization of citrus virologists conference proceedings (1957–2010)
Gong P, Tan H, Zhao S, Li H, Liu H, Ma Y, Zhang X, Rong J, Fu X, Lozano-Duran R, Li F, Zhou X (2021) Geminiviruses encode additional small proteins with specific subcellular localizations and virulence function. Nat Commun 12:4278
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Haible D, Kober S, Jeske H (2006) Rolling circle amplification revolutionizes diagnosis and genomics of geminiviruses. J Virol Methods 135:9–16
Hasanvand V, Heydanejad J, Massumi H, Kleinow T, Jeske H, Varsani A (2020) Isolation and characterization of a novel geminivirus from parsley. Virus Res 286:198056
Hesketh EL, Saunders K, Fisher C, Potze J, Stanley J, Lomonossoff GP, Ranson NA (2018) The 3.3 Å structure of a plant geminivirus using cryo-EM. Nat Commun 9:2369
Hipp K, Grimm C, Jeske H, Bottcher B (2017) Near-atomic resolution structure of a plant geminivirus determined by electron cryomicroscopy. Structure 25:1303–1309
Liang P, Navarro B, Zhang Z, Wang H, Lu M, Xiao H, Wu Q, Zhou X, Di Serio F, Li S (2015) Identification and characterization of a novel geminivirus with a monopartite genome infecting apple trees. J Gen Virol 96:2411–2420
Loconsole G, Saldarelli P, Doddapaneni H, Savino V, Martelli GP, Saponari M (2012) Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family Geminiviridae. Virology 432:162–172
Lu QY, Wu ZJ, Xia ZS, Xie LH (2015) Complete genome sequence of a novel monopartite geminivirus identified in mulberry (Morus alba L.). Arch Virol 160:2135–2138
Lu QY, Ma Y, Smith WK, Yu J, Cheng YY, Zhang P, Han TT (2021) The identification of Tautoneura mori as the vector of mulberry crinkle leaf virus and the infectivity of infectious clones in mulberry. Phytopathology. https://doi.org/10.1094/PHYTO-1003-1021-0094-R
Ma Y, Navarro B, Zhang Z, Lu M, Zhou X, Chi S, Di Serio F, Li S (2015) Identification and molecular characterization of a novel monopartite geminivirus associated with mulberry mosaic dwarf disease. J Gen Virol 96:2421–2434
Maclot F, Candresse T, Filloux D, Malmstrom CM, Roumagnac P, van der Vlugt R, Massart S (2020) Illuminating an ecological Blackbox: using high throughput sequencing to characterize the plant virome across scales. Front Microbiol 11:578064
Muhire B, Martin DP, Brown JK, Navas-Castillo J, Moriones E, Zerbini FM, Rivera-Bustamante R, Malathi VG, Briddon RW, Varsani A (2013) A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158:1411–1424
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Pringle CR (1999) Virus taxonomy–1999. The universal system of virus taxonomy, updated to include the new proposals ratified by the International Committee on Taxonomy of Viruses during 1998. Arch Virol 144:421–429
Qiu Y, Zhang S, Yu H, Xuan Z, Yang L, Zhan B, Murilo Zerbini F, Cao M (2020) Identification and characterization of two novel geminiviruses associated with paper mulberry (Broussonetia papyrifera) leaf curl disease. Plant Dis 104:3010–3018
Rojas MR, Macedo MA, Maliano MR, Soto-Aguilar M, Souza JO, Briddon RW, Kenyon L, Rivera Bustamante RF, Zerbini FM, Adkins S, Legg JP, Kvarnheden A, Wintermantel WM, Sudarshana MR, Peterschmitt M, Lapidot M, Martin DP, Moriones E, Inoue-Nagata AK, Gilbertson RL (2018) World management of geminiviruses. Annu Rev Phytopathol 56:637–677
Roossinck MJ, Martin DP, Roumagnac P (2015) Plant virus metagenomics: advances in virus discovery. Phytopathology 105:716–727
Saunders K, Richardson J, Lawson DM, Lomonossoff GP (2020) Requirements for the packaging of geminivirus circular single-stranded DNA: effect of DNA length and coat protein sequence. Viruses 12:1235
Siddell SG, Walker PJ, Lefkowitz EJ, Mushegian AR, Dutilh BE, Harrach B, Harrison RL, Junglen S, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Nibert ML, Rubino L, Sabanadzovic S, Simmonds P, Varsani A, Zerbini FM, Davison AJ (2020) Binomial nomenclature for virus species: a consultation. Arch Virol 165:519–525
Stover BC, Muller KF (2010) TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform 11:7
Vaghi Medina CG, Teppa E, Bornancini VA, Flores CR, Marino-Buslje C, Lopez Lambertini PM (2017) Tomato apical leaf curl virus: A novel, monopartite geminivirus detected in tomatoes in Argentina. Front Microbiol 8:2665
Varsani A, Martin DP, Navas-Castillo J, Moriones E, Hernandez-Zepeda C, Idris A, Murilo Zerbini F, Brown JK (2014) Revisiting the classification of curtoviruses based on genome-wide pairwise identity. Arch Virol 159:1873–1882
Varsani A, Navas-Castillo J, Moriones E, Hernandez-Zepeda C, Idris A, Brown JK, Murilo Zerbini F, Martin DP (2014) Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch Virol 159:2193–2203
Varsani A, Roumagnac P, Fuchs M, Navas-Castillo J, Moriones E, Idris A, Briddon RW, Rivera-Bustamante R, Murilo Zerbini F, Martin DP (2017) Capulavirus and Grablovirus: two new genera in the family Geminiviridae. Arch Virol 162:1819–1831
Yang Z, Zhang L, Zhao J, Li T, Liu Q, Cao M, Zhou Y (2020) First report of citrus chlorotic dwarf-associated virus on Pomelo in Nakhon, Thailand. Plant Dis 104:1262–1263
Zerbini FM, Briddon RW, Idris A, Martin DP, Moriones E, Navas-Castillo J, Rivera-Bustamante R, Roumagnac P, Varsani A, Ictv Report C (2017) ICTV virus taxonomy profile: Geminiviridae. J Gen Virol 98:131–133
Zhang R, Wu X, Jiang X, Wu X, Luan X, Cheng X (2020) Molecular characterization of common bean curly stunt virus: a novel recombinant geminivirus in China. Arch Virol 165:257–260
Zhang S, Shen P, Li M, Tian X, Zhou C, Cao M (2018) Discovery of a novel geminivirus associated with camellia chlorotic dwarf disease. Arch Virol 163:1709–1712
Zhang W, Olson NH, Baker TS, Faulkner L, Agbandje-McKenna M, Boulton MI, Davies JW, McKenna R (2001) Structure of the maize streak virus geminate particle. Virology 279:471–477
Zhou Y, Zhang Y, Liu Y, Chen H, Li T, Zhou C (2017) Distribution and molecular characterization of citrus chlorotic dwarf-associated virus in China. Australas Plant Pathol 46:227–229
Acknowledgements
JML is supported by the European Union (ERDF), the Conseil Régional de La Réunion, and CIRAD.
Funding
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling Editor: Sead Sabanadzovic.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
705_2021_5309_MOESM1_ESM.xlsx
Supplementary Data 1 Pairwise identity values for the genomes of viruses in the genera Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus, determined using SDT v1.2 [25] (XLSX 41 KB)
Rights and permissions
About this article

Cite this article
Roumagnac, P., Lett, JM., Fiallo-Olivé, E. et al. Establishment of five new genera in the family Geminiviridae: Citlodavirus, Maldovirus, Mulcrilevirus, Opunvirus, and Topilevirus. Arch Virol 167, 695–710 (2022). https://doi.org/10.1007/s00705-021-05309-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-05309-2