
Abstract
Bovine viral diarrhea virus (BVDV) is a major pathogen worldwide, causing significant economic losses to the livestock sector. In Uruguay, BVDV seroprevalence at the farm level is >80%. In this work, 2546 serum, blood or tissue samples collected from animals suspected of being affected by BVD between 2015 and 2017 were analyzed by reverse transcription PCR and sequencing. Analysis of the BVDV genomic regions 5’UTR/Npro, Npro and E2 revealed that BVDV-1a, 1i and 2b circulate in the country, with BVDV-1a being the most prevalent subtype. Population dynamics studies revealed that BVDV-1a has been circulating in our herds since ~1990. This subtype began to spread and evolve, accumulating point mutations at a rate of 3.48 × 10−3 substitutions/site/year, acquiring specific genetic characteristics that gave rise to two local genetic lineages of BVDV-1a. These lineages are divergent from those circulating worldwide, as well as the vaccine strain currently used in Uruguay. The most notable differences between field and vaccine strains were found in the E2 glycoprotein, suggesting that the amino acid substitutions could result in failure of cross-protection/neutralization after vaccination. This is the first study that compares Uruguayan BVDV field and vaccine strains with other BVDV strains from throughout the world. The results obtained in this study will be very useful for developing a suitable immunization program for BVDV in Uruguay by identifying local field strains as candidates for vaccine development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bovine viral diarrhea (BVD) is a disease of economic importance worldwide that causes a wide variety of clinical signs and reproductive failure in cattle [1]. The etiological agent, bovine viral diarrhea virus (BVDV), induces reproductive disorders (embryonic death, abortion, reduced fertility), immunosuppression, respiratory problems, diarrhea, and persistently infected (PI) calves that are immunotolerant to the virus [1,2,3]. According to a review by Yarnall and Thrusfield published in 2017, the economic impact of BVDV can vary between 0 to 710 dollars per cow per year. This range considers the subclinical disease, generally observed in herds with endemic BVDV infection, as well as epidemic or acute clinical infections that are seen more frequently in naive herds [4].
BVDV belongs to the genus Pestivirus in the family Flaviviridae, with a single-stranded positive-sense RNA genome of approximately 12.3 kb in length flanked at both ends by untranslated regions (UTRs). The only open reading frame (ORF) of BVDV encodes a single polyprotein that is cleaved co- and post-translationally into 11-12 structural and non-structural proteins, including Npro, C, Erns, E1, E2, p7, NS3 (NS2-NS3), NS4A, NS4B, NS5A, and NS5B [5].
There are three BVDV species currently recognized by the International Committee of Taxonomy Virus (ICTV), namely Pestivirus A, B and H, which include BVDV-1, BVDV-2, and HoBi-like pestivirus, respectively [6, 7].
Previous studies using BVDV nucleotide sequences of the 5’UTR, Npro and E2 genomic regions have revealed that BVDV-1 was the first to emerge, around 1802, followed by HoBi-like pestivirus in 1880 and BVDV-2 in 1890 [7].
BVDV-1 is the most genetically diverse, and at least 21 subtypes (BVDV-1a to BVDV-1u) have been described [8]. The greatest diversity of BVDV-1 subtypes has been described in strains circulating in Europe, China and Turkey [9]. In contrast, for BVDV-2, only four subtypes (BVDV-2a to BVDV-2d) have been described, and subtype 2a seems to predominate on all continents [9,10,11,12,13].
To study the genetic diversity of BVDV, different genomic regions have been used, including the 5’UTR, the membrane glycoprotein E2, the autoprotease Npro, and the protease NS3 [10, 14,15,16]. Currently, the 5’UTR is the main genomic region used for this purpose. Analysis of this genomic region has allowed the demarcation of BVDV species. However, due to its short length and high nucleotide conservation level, BVDV-1 subtypes usually cannot be easily defined, sometimes leading to incorrect subtype assignment [17, 18]. The use of 5’UTR/Npro allows proper assignment of BVDV strains to the different species and subtypes of BVDV [19]. Alternatively, the E2 glycoprotein, which is 1122 nucleotides in length, has been also used for this purpose. E2 is a less-conserved genomic region, and the most variable region of E2 is the amino-terminal end. The DA and DB domains of the E2 protein (residues 4-87 and 88-164, respectively), are the most exposed on the virus surface and therefore contain the main antigenic sites of the virion [20, 21]. Due to its variability, phylogenetic studies carried out with E2 have shown higher resolution than those based on other genomic regions. Additionally, comparative analysis of field and vaccine strains has been useful for discovering strains that are capable of escaping neutralizing antibodies [22].
BVDV-1 has a global distribution, while BVDV-2 seems to have a more restricted geographic distribution [9]. In South America, BVDV-1 predominates, with BVDV-1b being the most frequent subtype [9]. BVDV-1a is the most abundant subtype in Brazil, and it predominated in Argentina from 1984 to 1999 [23, 24]. There is a common belief that, due to the selective pressure produced by the BVDV-1a vaccine, there was a shift towards subtype BVDV-1b. Currently, BVDV-1b appears to be the most prevalent subtype in Argentina and is currently diversifying into a new group named “Arg1b” [25].
In our previous work, we reported that BVDV-1 and BVDV-2 circulate in Uruguayan herds, with BVDV-1a being the most frequent subtype [19]. The study of genetic variability among Uruguayan strains provides information to improve the diagnosis of the disease and represents a starting point for identifying local field strains for development of protective vaccines using homologous strains. In this work, as part of a national animal health project, we conducted a broad study of the molecular epidemiology of BVDV in Uruguay and deeper evolutionary analysis of BVDV-1a strains, the most prevalent subtype.
The results of this work will help to better understand the epidemiology, evolution and genetic variability of Uruguayan strains of BVDV and will provide tools to strengthen prevention and control strategies.
Materials and methods
Uruguayan BVDV sequences
Sample collection
From March 2015 to December 2017, a total of 2546 serum, blood and tissue samples were collected at the “Plataforma de Salud Animal” of the “Instituto Nacional de Investigación Agropecuaria”. Samples were obtained from calves with and without diarrhea, aborted fetuses, and heifers from farms where abortions had been reported. Samples were submitted to the molecular virology laboratory at the “Centro Universitario Salto, Universidad de la República”, and stored at -20°C until testing.
RNA extraction, reverse transcription and sample screening by real-time PCR
Viral RNA was extracted from all samples using a QIAamp cador Pathogen Mini Kit (QIAGEN, Germany) following the instructions specified by the manufacturer. In blood and serum samples, the protocol “Purification of pathogenic nucleic acids from fluid samples” was followed. Tissue samples were pretreated according to the “Pretreatment T1” protocol as described in the instructions. Reverse transcription was carried out using random primers and SuperScript II enzyme (Invitrogen, USA). For the detection of BVDV-1, BVDV-2 and HoBi-like pestivirus, a real-time PCR assay targeting 207 bp of the 5’UTR region of BVDV was performed using primers BVDV190F and V326 and the Taq- Man® probe TQ-Pesti as described by Hoffman et al. and Gaede et al., respectively, and latter modified by Maya et al. [19, 26, 27]. All real-time PCR reactions were performed using a SensiMix II Probe Kit (Bioline Reagents Ltd.) and a Rotor-Gene Q instrument (QIAGEN) following the manufacturer’s recommendations.
Amplification by conventional PCR
A fragment of 207 bp of the 5’UTR was amplified from real-time-PCR-positive samples, using the same primer pair that was used for the real-time PCR. Furthermore, a fragment of 428 bp of the Npro genomic region was amplified as described by Maya et al. [19]. A third amplicon of 606 bp belonging to the carboxyl-terminal region of the E1 glycoprotein (215 bp) and the amino-terminal region of the glycoprotein E2 (391 bp) was amplified using primers B11 and B32 with PCR conditions described by Couvreur et al. [28]. The resulting amplicons were sequenced by Macrogen (Seoul, South Korea) in an ABI3730XL Genetic Analyzer (Applied Biosystems, CA, USA).
Phylogenetic analysis
All of the sequences were edited using SeqMan Software (DNASTAR Lasergene) and aligned using the Clustal W algorithm in MEGA version 6.06 [29]. Genotype assignment was done by phylogenetic analysis of Uruguayan 5’UTR and Npro sequences concatenated into a fragment of 607 bp (5’UTR and Npro sequences overlap by 28 bp) and named “5’UTR/Npro”, along with sequences from Uruguayan BVDV strains published by Maya et al. (n = 14) and those of representative BVDV-1, BVDV-2 and HoBi-like pestivirus strains retrieved from the GenBank database. Sequences from Uruguayan strains are listed in Table 1. For samples from which only the 5’UTR region could be amplified, the subtype was determined using the NCBI BLAST tool [30].
Analysis of the genetic variability of BVDV-1a subtypes was performed using the coding region of Npro sequences (410 bp) and the amino-terminal E2 nucleotide sequences (391 bp) from Uruguayan strains and BVDV-1a strains from other countries based on the review by Yesilbag et al. [9], using sequences available in the GenBank database. The sequences used in these studies are summarized in Online Resources 1 and 2.
In all phylogenetic analyses, a reference sequence of border disease virus (BDV) was used as out-group. The model of nucleotide substitution that best fit the different datasets (GTR + gamma) was selected using the jModelTest program according to the Akaike information criterion (AIC; Akaike, 1974) [31]. Phylogenetic trees were constructed by the maximum-likelihood (ML) method, and statistical significance was evaluated by the bootstrap method (1,000 replicates) in MEGA version 6.06 [29].
Sequence analysis of Uruguayan BVDV-1a strains at the nucleotide and amino acid levels
The genetic distances of nucleotide and amino acid sequences were determined for the Uruguayan BVDV-1a strains and between the Uruguayan BVDV-1a strains and the NADL strain using Npro sequences (410 bp, and 136 amino acids) and those of E2 (391 bp and 131 amino acids), using MEGA version 6.06 [29].
The amino acid substitutions found in glycoprotein E2 of the Uruguayan BVDV-1a strains and the NADL strain were mapped to the crystallographic structure of E2 determined by El Omari et al. [20] (PDB ID: 2Y2Q) using PyMOL [32].
Population dynamics analysis of Uruguayan BVDV-1a
Before performing molecular clock analysis, the TempEst program was used to determine whether the sequences used in this work showed a temporal structure [33]. The time of the most recent common ancestor (tMRCA), the evolutionary rate of the 5’UTR/Npro (607 bp) of the Uruguayan BVDV-1a strains, and their demographic history were jointly estimated using the BEAST v1.7.5 package (available online: http://tree.bio.ed.ac.uk/software/beast/) [34]. Bayesian analysis was conducted under relaxed molecular clock models with uncorrelated lognormal (UCLN) and exponential (UCED) distributions and the strict molecular clock model. The three molecular clock models were evaluated in combination with the constant size, exponential growth, expansion growth and Bayesian skyline coalescent tree prior using Bayes Factors. Markov chain Monte Carlo (MCMC) simulations were run for 200 million generations, and the results were visualized using the Tracer v1.5.0 program (available from http://beast.bio.ed.ac.uk/Tracer), excluding the initial 10% of the run as burn-in. The effective sample size (ESS) values were checked to evaluate the convergence of the analysis, accepting only values higher than 200 for all of the parameters. The effective number of infections was represented graphically using Tracer v1.5.0.
Results
Genetic diversity of Uruguayan BVDV strains based on analysis of 5′UTR/Npro sequences
Twenty-three samples were positive for BVDV by real-time PCR. These 23 samples, added to the 16 samples previously obtained by our group, gave a total of 39 samples that were positive for BVDV (Table 1) [19].
In five of the 23 samples, only the 5’UTR region could be amplified, and the subtype was determined using the NCBI BLAST tool, with three samples being characterized as BVDV-1a, and two samples as BVDV-2b. In our previous work, two of the 16 positive samples were classified as BVDV-1a by BLAST, so we obtained a total of five BVDV-1a and two BVDV-2b isolates (Table 1) [19].
From the remaining 18 positive samples, the 5’UTR and Npro genomic regions could be successfully amplified, sequenced, edited and subsequently concatenated to 5’UTR/Npro fragments. Adding these 18 sequences to those described by Maya et al. [19] (n = 14), a total of 32 Uruguayan 5’UTR/Npro sequences from strains from 2014-2017 were used for phylogenetic analysis and subsequent studies.
According to the results obtained from jModelTest, the best model of nucleotide substitution that described our dataset was GTR + gamma.
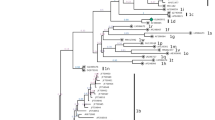
The 32 samples were genotyped by performing a phylogenetic analysis based on a 607-bp fragment of the 5’UTR/Npro region (Fig. 1). We found that 29 Uruguayan strains belonged to the species Pestivirus A (BVDV-1), with 91% statistical support, and three belonged to the species Pestivirus B (BVDV-2), with 100% support. Inside the BVDV-1 cluster, 28 of the 29 samples belonged to the subtype BVDV-1a, and one sample belonged to the subtype BVDV-1i. All BVDV-2 strains were assigned to BVDV-2b subtype.

Phylogenetic analysis of the 5’UTR/Npro region of BVDV strains. Uruguayan strains are indicated by black dots. BVDV-1a, 1i and 2b are highlighted in gray and indicated on the right side of the figure. Numbers at the branches of the trees are bootstrap values. A BDV (border disease virus) sequence was included in the analysis as an out-group
The sample name, viral species/subtype, geographic location (department) and GenBank accession number of the 5 ‘UTR/Npro, 5´UTR and E1-E2 genomic regions are summarized in Table 1.
Phylogenetic analysis shows that the Uruguayan BVDV-1a strains (n = 28) were subdivided into two clades, named BVDV-1a lineage 1 UY and BVDV-1a lineage 2 UY (Figure 1). BVDV-1a lineage 1 UY was the main clade, constituted by 24 BVDV-1a sequences. This lineage formed a group supported by an 81% bootstrap value, along with five Brazilian strains (LV60-53/13, LV85-59/13, LV17-09/13, SV663/00 and LV86-80/13).
The smaller Uruguayan subgroup, designated as BVDV-1a lineage 2 UY, with 99% bootstrap support, is formed by the remaining four Uruguayan sequences (see Fig. 1).
The Uruguayan strain 436FaUY/052014 of the BVDV-1i subtype clusters together with the Brazilian strain ACM/BR/2016 and with strains from the United Kingdom. Within this group, the strains from the United Kingdom grouped separately from the two Latin American strains, with 99% statistical support.
The three Uruguayan strains of the BVDV-2b subtype were divided into two groups: strain 439RvUY/082014 grouped with Brazilian strains of this subtype, and strains 2391UYRN/2016 and 2769UYRN/2016 together formed a separate group supported by a bootstrap value of 99%.
Analysis of the Npro genomic region
Nucleotide and amino acid distances among Uruguayan BVDV-1a strains
The 24 sequences of BVDV-1a lineage1 UY had a nucleotide sequence divergence of 4.4% (18/410) and an amino acid sequence divergence of 2.9% (4/136). The 4 sequences of BVDV-1a lineage 2 UY had a nucleotide sequence divergence of 2.9% (12/410) an amino acid sequence divergence of 0.7% (1/136). These two Uruguayan lineages diverge in 11.5% of their nucleotides (47/410), and 5.2% (7/136) of their amino acids (Table 2).
Both Uruguayan lineages are genetically divergent with respect to the NADL vaccine strain. BVDV-1a lineage 1 UY and the NADL strain diverge in 11% of their nucleotides (45/410) and 5.1% of their amino acids (7/136). The BVDV-1a lineage 2 UY diverges from the NADL strain in 9.2% of its nucleotides (38/410) and 5.9% of its amino acids (8/136) (Table 2). In the Npro protein, the two Uruguayan lineages have the residues N/E30I39K75F110, which differ from those in the NADL strain, which has D30V39R75S110 (Table 3a).
Phylogenetic analysis of the Npro genomic region
A second phylogenetic analysis (Fig. 2) was performed with 143 nucleotide sequences of the Npro protease (410 bp) from the BVDV-1a subtype isolates from Uruguay (n = 28) and strains from Austria, Brazil, China, Canada, the United Kingdom and the United States of America, (Online resource 1) selected based on a review by Yesilbag et al. [9].

Phylogenetic analysis of Npro sequences of BVDV-1a strains. Uruguayan BVDV-1a strains are indicated by dots. The UK clade is indicated by triangles, the CA clade by squares, and the BR clade by rhombuses. A BVD (border disease virus) sequence was included in the analysis as an out-group
The results of these studies revealed that several groups of BVDV-1a strains can be observed (Fig. 2). The Uruguayan strains of lineage BVDV-1a showed the same topology when using the 5′UTR/Npro or Npro region for analysis (see Fig. 1 and 2). Strains of BVDV-1a lineage 1 UY were grouped with 92% statistical support with the five Brazilian strains and the Chinese strain G5S. Strains of BVDV-1a lineage 2 UY grouped together with a bootstrap value of 88%. Strains from the United Kingdom formed several groups (named UK1 to UK8). The Brazilian strains were separate from Uruguayan lineage 1 UY and formed a group (BR clade). The same was observed for the Canadian strains, which formed a group called the “CA clade” (Online Resource 1).
Population dynamics analysis of Uruguayan BVDV-1a isolates
To better characterize the Uruguayan BVDV1a strains, and their evolutionary and demographic history, we estimated their tMRCA and molecular clock. The nucleotide substitution model that best fit our data was GTR + gamma. The results obtained using Tempest showed that the 5’UTR/Npro genomic region had a temporal structure, and therefore we could proceed to other estimates.
The results of these analyses revealed that this subtype began to spread in Uruguay in 1990 at a substitution rate of 3.48 × 10−3 substitutions/site/year (Table 4).
Analysis of the E1-E2 genomic region
Nucleotide and amino acid distances between Uruguayan BVDV-1a strains
E1-E2 nucleotide sequences (603 bp) were obtained for 15 Uruguayan strains of the BVDV-1a subtype, 14 of BVDV-1a lineage 1 UY, and one of the BVDV-1a lineage 2 UY (438TboUY/042014), (see Table 1). The amplified region of E2 (391 bp, and 131 amino acids of a total of 373) is shown in white in Fig. 4a and b and corresponds to domains DA (amino acids 4-87) and partly to domain DB (amino acids 88-164).
The E1-E2 region of the BVDV-1a lineage 1 UY isolates showed a genetic diversity of 5.5% at the nucleotide level and 6% at the amino acid level (Table 2). BVDV-1a lineage 1 UY differed from BVDV-1a lineage 2 UY in 14.3% (86/603) of its nucleotides and 12.9% of its amino acids (Table 2). BVDV-1a lineage 1 UY has a nucleotide divergence from the NADL vaccine strain of 13.8%, and 15.4% at the amino acid level. When BVDV-1a lineage 2 UY (strain 438TboUY/042014) and the NADL strain were compared, a divergence of 14.8% and 13.4% at the nucleotide and amino acid level, respectively, was observed (Table 2).
The NADL strain differs from the two Uruguayan lineages of BVDV-1a at 11 amino acid positions in the E2 glycoprotein (Table 3b). Seven amino acid changes were found in beta sheets (aa 15, 20, 38, 49, 54, 68 and 81) and are shown as white spheres in Fig. 4b, and the four remaining residues were in the loops between the beta sheets (aa 71, 87, 88 and 90) and are shown as light gray spheres in Fig. 4b. El Omari et al. mapped a linear epitope of CSFV in the structure of glycoprotein E2 of BVDV from amino acid 83 to 90 in the DA and DB domains, in a loop between two beta sheets (light gray, Fig. 4a) [20]. Substitutions at amino acids 87, 88 and 90 found in Uruguayan lineages of BVDV-1a with respect to the NADL strain are part of this linear epitope (represented by light gray spheres in Fig. 4b). An immunodominant epitope has been mapped in BVDV E2 from amino acid 71 to 74 of E2 (light gray, Fig. 4a) [35]. A substitution at amino acid 71 of E2 was observed in most of the Uruguayan BVDV-1a strains (12 of 15) when compared to the NADL strain (represented by a dark gray sphere in Fig. 4b).
Uruguayan BVDV-1a lineages differ from each other by six non-synonymous substitutions in the E2 protein (aa 15, 38, 54, 68, 81 and 90), and substitutions at positions 38, 54, 68 and 81 of E2 were found only in the Uruguayan strains of BVDV-1a lineage 1 UY (Table 3b).
Phylogenetic analysis of the E2 genomic region
A third phylogenetic analysis was done using 33 nucleotide sequences from the amino-terminal region of the membrane glycoprotein E2 (391 bp) (see Fig. 3), including strains from the BVDV-1a subtype from Uruguay (n = 15) and strains from China, Japan, the United States of America and Argentina (Online Resource 2), selected on the basis of a review by Yesilbag et al. [9]. To date, no available and comparable sequences of the amino-terminal region of E2 from Austria, Brazil and the United Kingdom are available, so a phylogenetic tree of Npro and E2 could not be made with sequences from those countries. The Canadian sequences of E2 obtained by Chernick et al. [22] were too short (245 bp) to be included in the analysis. The results of these studies are shown in Figure 3.

Phylogenetic analysis of E2 nucleotide sequences of BVDV-1a strains. Uruguayan BVDV-1a strains are indicated by dots. A BDV (border disease virus) sequence was included in the analysis as an out-group
Strains of BVDV-1a lineage 1 UY clustered together with the Chinese strain G5S, with 95% bootstrap support, while BVDV-1a lineage 2 UY grouped with the Chinese strain GS24, with 94% bootstrap support (see Fig. 3). The BVDV-1a strains from Japan formed a separate group called the “JP group”, with 98% statistical support. Strains from Argentina did not cluster together, revealing a significant degree of genetic variability among them.
Discussion
BVD is one of the most important infectious diseases of livestock, causing significant economic losses to the livestock industry. In Australia, the beef and dairy industries are among the top three in the agricultural sector. In Australia, there are no mitigation programs for BVD, and this disease is second in importance to diseases carried by ticks. In tick-free areas, BVDV is the most important pathogen [36].
In Uruguay, cattle industry exports represent around 10% of the country’s gross domestic product [37]. There is a cattle population of around 12 million head, including beef and dairy breeds, distributed on 42,114 farms. The bovine population in Uruguay has remained practically unchanged since 1998, and it is suspected that BVDV is one of the factors that contribute to this situation, affecting pregnancy and procreation [38]. Research studies carried out in Uruguay in 2000 revealed that only 3% of farmers vaccinated cattle against BVDV. BVDV was found on 100% of the farms studied, and a seroprevalence of 67% was found in cattle [39]. These data were updated recently with similar values of seroprevalence at the farm level (98.8%) and higher seroprevalence at the herd level (approximately 80%, Dr. Federico Fernández, Ministerio de Ganadería, Agricultura y Pesca, MGAP, personal communication). Yarnall and Thrusfield estimated that farms endemically infected with BVDV could experience an economic impact ranging from 8.4 to 113 dollars per cow per year [4].
In Uruguay, there is no national program for the eradication of BVDV, and vaccination is not mandatory. The vaccines against BVDV available in Uruguay are listed in the national annual report to the World Organization for Animal Health (OIE); they are inactivated polyvalent vaccines that contain BVDV-1 and BVDV-2 [40]. Only one of the manufacturers states that the vaccine contains the NADL reference strain of the BVDV-1a subtype and the SV323 strain of the BVDV-2a subtype. No information is available on the viral subtypes included in the vaccines prepared by other manufacturers.
Many countries, such as Austria, Great Britain, Denmark, Finland, France, Germany, Ireland, Italy, the Netherlands, Norway, Scotland, Slovenia, Sweden and Switzerland have BVDV eradication programs in progress at the national or regional level [36]. In Switzerland, the virus has been successfully eradicated [41].
BVDV eradication can lead to a significant reduction of economic losses. To achieve this goal, a control policy should be implemented consisting in identification and removal of PI animals, identification of natural in utero infection of calves, continued surveillance of animals showing clinical signs characteristic of BVD, and an effective vaccination program [42, 43].
In our previous work, we found that BVDV-1 and BVDV-2 were circulating in Uruguayan herds with reproductive problems and that the most frequent subtype was BVDV-1a [19]. The current work extends the sampling and supports the molecular epidemiology data obtained previously, given that the BVDV-1a subtype continues to be the most frequent subtype in our country.
The geographical diversification of the Uruguayan strains of BVDV-1a described in our previous work became more evident when increasing the number of positive samples of this subtype. Phylogenetic analysis clearly showed two clades of Uruguayan BVDV-1a (Figs. 1, 2 and 3). These clades were named “BVDV-1a lineage 1 UY” and “BVDV-1a lineage 2 UY”. BVDV-1a lineage 1 UY included a larger number of Uruguayan strains (n = 24) and formed a clade together with some Brazilian strains and the GS5 strain isolated in China (Figs. 1 and 2). In contrast, BVDV-1a lineage 2 UY included fewer Uruguayan strains (n = 4) and only one foreign strain, GS24 of Chinese origin (Fig. 3).
According to the tMRCA estimate, the BVDV-1a subtype began circulating in Uruguay in 1990, approximately 27 years ago. Since then, this subtype began to spread and evolve, accumulating substitutions at a rate of 3.48 × 10−3 substitutions/site/year (s/s/y) in the 5’UTR/Npro region. In this way, the BVDV-1a subtype in Uruguay diversified by acquiring local specific genetic characteristics that gave rise to two BVDV-1a lineages, which are genetically different from the BVDV-1a strains isolated in other countries and from the vaccine strain NADL. The estimates of the substitution rates found in these analyses (3.48 ×10−3 s/s/y) are consistent with those reported in previous studies [22, 44].
The results of these studies revealed that both of the BVDV-1a lineages circulating in Uruguay were genetically distant from the NADL vaccine strain (see Figs. 1-3). In the Npro protease region, the two Uruguayan lineages differed from the NADL strain by four amino acid changes N/E30D, I39V, K75R, F110S (Table 3a).
In order to obtain more-detailed information on the divergence between the NADL vaccine strain and the Uruguayan BVDV-1a strains, we analyzed the amino-terminal region of glycoprotein E2. This region is more variable than the Npro region due to its role in the replicative cycle of the virus. The amino-terminal region of E2 is the most exposed region on the surface of the virus and therefore is the most antigenic region of this protein [20]. In the E2 region, most of the nucleotide changes are non-synonymous and lead to amino acid changes (see Table 2). In this region, 11 amino acid changes were found when comparing the two Uruguayan lineages of BVDV-1a and the NADL strain, revealing a significant degree of genetic variability among vaccine strain and strains circulating in Uruguay (Table 3b, Fig. 4). Substitutions at residues 71, 87, 88 and 90 found in the two BVDV-1a lineages circulating in Uruguay relative to the NADL strain are part of two epitopes of E2 [20, 35]. Therefore, these substitutions in glycoprotein E2 of the Uruguayan strains could lead to a protection/neutralization failure in animals vaccinated with the NADL strain. More studies will be needed to address this important issue.

Cartoon representation of the E2 monomer. a) The amplified region of the E2 glycoprotein is shown in white, and the rest of the monomer is in dark grey. Antigenic sites mapped in classical swine fever virus and BVDV are shown in light grey and indicated by squares. b) Cartoon representation of the E2 monomer rotated about the y-axis 180 degrees counterclockwise. The 11 amino acid substitutions found in the Uruguayan strains of the BVDV-1a subtype are represented as spheres colored light grey if located in a known antigenic site. Otherwise, they are shown in white
In addition to the mutations described above, the Uruguayan sequences from BVDV-1a lineage 1 UY had a unique fingerprint T38V54T68E81, in glycoprotein E2, which differentiates them from strains of BVDV-1a lineage 2 UY (M38M54I68K81), the NADL strain (K38M54I68K81), and also the rest of the strains of the BVDV-1a subtype included in these analyses (Table 3b, shown in white, Fig. 4a and b). Although the amino acid changes at residues 38, 68 and 81 were not within previously reported antigenic sites, they might cause changes in the properties of the E2 protein. The mutation at residue 54 of BVDV-1a lineage 1 UY resulted in a smaller R group in the valine residue than in the methionine residue shared by the NADL vaccine strain and members of BVDV-1a lineage 2 UY. More studies are needed to determine whether these amino acid changes are relevant for the fitness of the Uruguayan strains with this amino acid fingerprint.
The greatest diversity of BVDV-1 subtypes has been described in Europe. The BVDV-1i subtype was first described in the United Kingdom in 1999. It appeared to be restricted to that country until 2014, when it was found in Uruguay, and in 2016, it was also found in Brazil [19, 45, 46]. The strains from the United Kingdom grouped together with 99% statistical support and were separated from the strains from Brazil and Uruguay (Fig. 1). Strains isolated in South America have a close genetic relationship with each other in the Npro region and a more distant genetic distance with strains isolated in the United Kingdom [46].
In contrast to the BVDV-1, the BVDV-2 is not common in South America. However, BVDV-2b is the predominant subtype in Brazil [9]. The two Uruguayan strains belonging to the BVDV-2b subtype (2391UYRN/2016 and 2769UYRN/2016) clustered together, separated from most of the other strains of this subtype, including the Uruguayan strain 439RvUY/082014. More studies will be needed in order to observe if genetic diversification is taking place within the BVDV-2b genotype in Uruguay.
The relationship between the Brazilian, British, Chinese and Uruguayan strains (BVDV-1a, 1i and BVDV-2b) could probably be explained by the movement of the virus between these countries through commercial relationships in the livestock sector.
In summary, BVDV is a major health problem in Uruguayan herds, affecting production and reproduction. Appropriate prevention measures should be applied in order to control this pathogen and reduce the economic losses it causes. This study provides information about the molecular epidemiology of BVDV in our country. The BVDV-1a subtype is the most prevalent in Uruguay, and it has evolved over three decades, acquiring local characteristics that make it different from the NADL vaccine strain.
The Uruguayan subtype BVDV-2b should be included in the vaccine formulation, even though only a few isolates of this subtype were found. BVDV-2b also seems to be diverging in the Uruguayan territory, so surveillance of BVDV needs to be continued in order to have a clear picture of the behavior of this viral subtype in our country.
The results of these studies suggest that, to achieve adequate prophylaxis, it might be adequate to reformulate existing vaccines to contain the two types of BVDV and the subtypes BVDV-1a and BVDV-2b.
Additional in-depth studies are needed to investigate the antigenic characteristics and determine the complete genome sequences of Uruguayan BVDV strains in order to select local isolates as candidates for developing a suitable protective vaccine for our herds.
References
Houe H (1999) Epidemiological features and economic importance of bovine virus diarrhoea virus (BVDV) infections. Vet Microbiol 64:89–107
Baker JC (1987) Bovine viral diarrhea virus: a review. J Am Vet Med Assoc 190:1449–1458
Baker JC (1995) The clinical manifestations of bovine viral diarrhea infection. Vet Clin N Am Food Anim Pract 11:425–445
Yarnall MJ, Thrusfield MV (2017) Engaging veterinarians and farmers in eradicating bovine viral diarrhoea: a systematic review of economic impact. Vet Rec 181(13):347. https://doi.org/10.1136/vr.104370
Meyers G, Thiel HJ (1996) Molecular characterization of pestiviruses. Adv Virus Res 47:53–118
International Committee on Taxonomy of Viruses ICTV. https://talk.ictvonline.org/
Liu L, Xia H, Wahlberg N, Belák S, Baule C (2009) Phylogeny, classification and evolutionary insights into pestiviruses. Virology 385:351–357. https://doi.org/10.1016/j.virol.2008.12.004
Ridpath JF, Fulton RW, Kirkland PD, Neil JD (2010) Prevalence and antigenic differences observed between bovine viral diarrhea virus subgenotypes isolated from cattle in Australia and feedlots in the southwestern United States. J Vet Diagn Investig 22:184–191. https://doi.org/10.1177/104063871002200203
Yesilbag K, Alpay G, Becher P (2017) Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses 9(6):128. https://doi.org/10.3390/v9060128
Vilcek S, Herring AJ, Herring JA, Nettleton PF, Lowings JP, Paton DJ (1994) Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Arch Virol 136:309–323
Flores EF, Ridpath JF, Weiblen R, Vogel FS, Gil LH (2002) Phylogenetic analysis of Brazilian bovine viral diarrhea virus type 2 (BVDV-2) isolates: Evidence for a subgenotype within BVDV-2. Virus Res 87:51–60
Peterhans E, Bachofen C, Stalder H, Schweizer M (2010) Cytopathic bovine viral diarrhea viruses (BVDV): emerging pestiviruses doomed to extinction. Vet Res. https://doi.org/10.1051/vetres/2010016
Jenckel M, Hӧper D, Schirrmeier H, Reimann I, Goller KV, Hoffmann B, Beer M (2014) Mixed triple: allied viruses in unique recent isolates of highly virulent type 2 bovine viral diarrhea virus detected by deep sequencing. J Virol 88:6983–6992. https://doi.org/10.1128/JVI.00620-14
Ridpath JF, Bolin SR, Dubovi EJ (1994) Segregation of bovine viral diarrhea virus into genotypes. Virology 205:66–74. https://doi.org/10.1006/viro.1994.1620
Becher P, Orlich M, Shannon AD, Horner G, Konig M, Thiel HJ (1997) Phylogenetic analysis of pestiviruses from domestic and wild ruminants. J Gen Virology 78:1357–1366. https://doi.org/10.1099/0022-1317-78-6-1357
Weber MN, Streck AF, Silveira S, Mósena ACS, da Silva MS, Canal CW (2015) Homologous recombination in pestiviruses: identification of three putative novel events between different subtypes/genogroups. Infect Genet Evol 30:219–224. https://doi.org/10.1016/j.meegid.2014.12.032
Xia H, Liu L, Wahlberg N, Baule C, Belák S (2007) Molecular phylogenetic analysis of bovine viral diarrhea virus, a Bayesian approach. Virus Res 130:53–62. https://doi.org/10.1016/j.virusres.2007.05.017
Booth RE, Thomas CJ, El-Attar LM, Gunn G, Brownlie J (2013) A phylogenetic analysis of Bovine Viral Diarrhoea Virus (BVDV) isolates from six different regions of the UK and links to animal movement data. Vet Res 44(1):43–56. https://doi.org/10.1186/1297-9716-44-43
Maya L, Puentes R, Reolón E, Acuña P, Riet F, Rivero R, Cristina J, Colina R (2016) Molecular diversity of bovine viral diarrhea virus in Uruguay. Arch Virol 1613:529–535. https://doi.org/10.1007/s00705-015-2688-4
El Omari K, Iourin O, Harlos K, Grimes JM, Stuart DI (2013) Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Rep 3(1):30–35. https://doi.org/10.1016/j.celrep.2012.12.001
Neill JD (2013) Molecular biology of bovine viral diarrhea virus. Biologicals 41(1):2–7. https://doi.org/10.1016/j.biologicals.2012.07.002
Chernick A, Godson DL, van der Meer F (2014) Metadata beyond the sequence enables the phylodynamic inference of bovine viral diarrhea virus type 1a isolates from Western Canada. Infect Genet Evol 28:367–374. https://doi.org/10.1016/j.meegid.2014.01.003
Jones LR, Zandomeni R, Weber EL (2001) Genetic typing of bovine viral diarrhea virus isolates from Argentina. Vet Microbiol 81:367–375
Weber MN, Silveira V, Machado G, Groff FHS, Mósena ACS, Budaszewski RF, Dupont PM, Corbellini LG, Canal CW (2014) High frequency of bovine viral diarrhea virus type 2 in Southern Brazil. Virus Res 191:117–124. https://doi.org/10.1016/j.virusres.2014.07.035
Pecora A, Malacari DA, Ridpath JF, Perez Aguirreburualde MS, Combessies G, Odeon AC, Romera SA, Golemba MD, Wigdorovitz A (2014) First finding of genetic and antigenic diversity in 1b-BVDV isolates from Argentina. Res Vet Sci 96:204–212. https://doi.org/10.1016/j.rvsc.2013.11.004
Hoffmann B, Depner K, Schirrmeier H, Beer M (2006) A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J Virol Methods 136(1–2):200–209. https://doi.org/10.1016/j.jviromet.2006.05.020
Gaede W, Reiting R, Schirrmeier H, Depner KR, Beer M (2005) Detection and species-specific differentiation of pestiviruses using real-time RT-PCR. Berl Munch Tierarztl Wochenschr 118:113–120
Couvreur B, Letellier C, Collard A, Quenon P, Dehan P, Hamers C, Pastoret PP, Kerkhofs P (2002) Genetic and antigenic variability in bovine viral diarrhea virus (BVDV) isolates from Belgium. Virus Res 85:17–28
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. https://doi.org/10.1093/molbev/msn083
PyMOL Molecular graphics system, version 2.0 Schrödinger, LLC
Rambaut A, Lam TT, Max Carvalho L, Pybus OG (2016) Exploring the temporal structure of heterochronous sequences using TempEst (formely Path-O-Gen). Virus Evol 2(1):vew007. https://doi.org/10.1093/ve/vew007
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973. https://doi.org/10.1093/molbev/mss075
Ridpath J (2013) Immunology of BVDV vaccines. Biologicals 41:14–19. https://doi.org/10.1016/j.biologicals.2012.07.003
Reichel MP, Lanyon SR, Hill FI (2018) Perspectives on current challenges and opportunities for Bovine Viral Diarrhoea VirusEradication in Australia and New Zeland. Pathogens 1:7–14. https://doi.org/10.3390/pathogens7010014
Ministerio de Ganaderia Agricultura y Pesca-Uruguay (MGAP)-Dirección de Estadística Agropecuaria (DIEA). http://www.mgap.gub.uy/unidad-organizativa/oficina-de-programacion-y-politicas-agropecuarias/publicaciones/anuarios-diea
Presidencia de la República Oriental del Uruguay-Plan Nacional de Investigación en sanidad animal-2009. http://archivo.presidencia.gub.uy/_web/MEM_2009/MGAP.pdf
Guarino H, Núñez A, Repiso MV, Gil A, Dargatz DA (2008) Prevalence of serum antibodies to bovine herpesvirus-1 and bovine viral diarrhea virus in beef cattle in Uruguay. Prev Vet Med 85:34–40. https://doi.org/10.1016/j.prevetmed.2007.12.012
World Organization for animal health (OIE). https://www.oie.int/wahis_2/public/wahid.php/Reviewreport/semestrial/reviewyear=2017&semester=0&wild=0&country=URY&this_country_code=URY&detailed=1
Presi P, Heim D (2010) BVD eradication in Switzerland—a new approach. Vet Microbiol 142:137–142. https://doi.org/10.1016/j.vetmic.2009.09.054
Al-Khaliyfa MA, Abuelzein EME, Gameel AA (2010) Identification of cattle persistently infected with BVDV by ear-notch testing in Saudi Arabia. Vet Rec 167(17):660–661. https://doi.org/10.1136/vr.c5270
Abuelzein EM, Al-Khaliyfa MJ, Gameel AA (2011) Natural in utero infection of neonatal calves with bovine viral diarrhoea virus on a large dairy farm in Saudi Arabia. Onderstepoort J Vet Res 78(1):318. https://doi.org/10.4102/ojvr.v78i1.318
Luzzago C, Ebranati E, Sassera D, Lo Presti A, Lauzi S, Gabanelli E, Ciccozzi M, Zehender G (2012) Spatial and temporal reconstruction of bovine viral diarrhea virus genotype 1 dispersion in Italy. Infect Genet Evol 12:324–331. https://doi.org/10.1155/2014/147145
Strong R, Errington J, Cook R, Ross-Smith N, Wakeley P, Steinbach F (2013) Increased phylogenetic diversity of bovine viral diarrhoea virus type 1 isolates in England and Wales since 2001. Vet Microbiol 162:315–320. https://doi.org/10.1016/j.vetmic.2012.09.006
Mósena AC, Weber MN, Cibulski SP, Silveira S, Silva MS, Mayer FQ, Canal CW (2017) Genomic characterization of a bovine viral diarrhea virus subtype 1i in Brazil. Arch Virol 1624:1119–1123. https://doi.org/10.1007/s00705-016-3199-7
Acknowledgements
This work was supported primarily by Grant FSA_1_2013_1_12915 ‘‘Estudio de la epidemiología molecular y prevalencia serológica del virus de la Diarrea Viral Bovina en rodeos de carne y leche en el Uruguay con el fin de seleccionar cepas para una vacuna protectora de alcance masivo’’, from ANII (Agencia Nacional de Investigación e Innovación). This work was also partially funded by Grants FSSA_X_2014_1_105696 “Causas y prevalencia de abortos y evaluación de índices reproductivos en rodeos bovinos lecheros con enfermedades abortivas en Uruguay”, and FMV_1_2014_1_104922 from ANII, and Grant PL-015 N-15156 “Estimación de la morbilidad y mortalidad de las enfermedades que afectan la cría de los terneros lecheros del Uruguay e identificación de los principales factores asociados a sus frecuencias” from INIA. We thank the PEDECIBA program. We also thank the staff at the PSA (Plataforma de Salud Animal) of INIA (Instituto Nacional de Investigación Agropecuaria).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Zhenhai Chen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Maya, L., Macías-Rioseco, M., Silveira, C. et al. An extensive field study reveals the circulation of new genetic variants of subtype 1a of bovine viral diarrhea virus in Uruguay. Arch Virol 165, 145–156 (2020). https://doi.org/10.1007/s00705-019-04446-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04446-z