
Abstract
The family Arteriviridae presently includes a single genus Arterivirus. This genus includes four species as the taxonomic homes for equine arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV), porcine respiratory and reproductive syndrome virus (PRRSV), and simian hemorrhagic fever virus (SHFV), respectively. A revision of this classification is urgently needed to accommodate the recent description of eleven highly divergent simian arteriviruses in diverse African nonhuman primates, one novel arterivirus in an African forest giant pouched rat, and a novel arterivirus in common brushtails in New Zealand. In addition, the current arterivirus nomenclature is not in accordance with the most recent version of the International Code of Virus Classification and Nomenclature. Here we outline an updated, amended, and improved arterivirus taxonomy based on current data. Taxon-specific sequence cut-offs are established relying on a newly established open reading frame 1b phylogeny and pairwise sequence comparison (PASC) of coding-complete arterivirus genomes. As a result, the current genus Arterivirus is replaced by five genera: Equartevirus (for EAV), Rodartevirus (LDV + PRRSV), Simartevirus (SHFV + simian arteriviruses), Nesartevirus (for the arterivirus from forest giant pouched rats), and Dipartevirus (common brushtail arterivirus). The current species Porcine reproductive and respiratory syndrome virus is divided into two species to accommodate the clear divergence of the European and American “types” of PRRSV, both of which now receive virus status. The current species Simian hemorrhagic fever virus is divided into nine species to accommodate the twelve known simian arteriviruses. Non-Latinized binomial species names are introduced to replace all current species names to clearly differentiate them from virus names, which remain largely unchanged.
Similar content being viewed by others


Introduction
In comparison to prokaryotic and eukaryotic taxonomy, viral taxonomy is still in its infancy. The most recent updates issued by the International Committee on Taxonomy of Viruses (ICTV) list <10,000 viruses that have been classified in <3,000 species [2, 3, 6, 7, 30]. The development and increasing adoption of metagenomic approaches in recent years have revealed that current measurements of global viral diversity are vast underestimates [8, 13]. For the foreseeable future, virus taxonomy will have to be revised continuously and sometimes substantially as new discoveries are made that challenge current taxonomic thinking [38]. Objective techniques, such as viral genome sequence-based alignments and comparison algorithms, and automatic tools (in silico taxonomy decision trees) are used increasingly for virus classification to circumvent virus culture bias (i.e., the acceptance and classification of only that minority of viruses that can be grown in the laboratory). Updating existing taxonomies is crucial for classification to become database-compliant and user-friendly, while not violating the Rules of the International Code of Virus Classification and Nomenclature (ICVCN), which also are updated continuously [4, 5, 30].
Here, we address the taxonomy of the family Arteriviridae (order Nidovirales). Arteriviruses have monopartite, single-stranded RNA genomes of positive polarity. Large nonstructural polyproteins are directly translated from these genomes, whereas additional structural proteins are typically translated from nested sets of subgenomic mRNAs [24]. We outline an updated arterivirus classification and nomenclature to incorporate recent discoveries and to address current shortcomings and inconsistencies for ease of application, clarity, and ICVCN compliance.
History of arterivirus taxonomy
The taxonomic history of equine arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV), porcine respiratory and reproductive syndrome virus (PRRSV), and simian hemorrhagic fever virus (SHFV)—currently the only accepted members of the family Arteriviridae—is complex. The reason for this complexity is in part because these viruses were discovered before the advent of modern molecular biological techniques. The isolation of EAV, the causative agent of equine viral arteritis, was reported in 1957 [15]; the isolation of LDV, which can cause disease in laboratory mice, was reported in 1963 [44]; and the isolation of SHFV, which causes simian hemorrhagic fever in Asian macaques, was reported in 1968 [52]. Even the isolation of PRRSV, the etiological agent of porcine reproductive and respiratory syndrome, in 1991 [57] (European/Lelystad “strain” or “type 1”) and 1992 [12, 19] (North American “strain” or “type 2”) predates the availability of routine genome sequencing. Consequently, initial classification of these viruses relied primarily on morphological features of their virions (Fig. 1), with subsequent reclassifications based on nucleotide sequences of genome fragments or, eventually, coding-complete or complete genomes generated from viruses isolated in vitro.

Structure of arterivirions. A) Electron micrograph of simian hemorrhagic fever virus particles inside of cellular vacuoles. B) Cartoon of a generalized simian arterivirus particle depicting all structural proteins. Arteriviruses from non-simian hosts, such as African pouched rat virus 1, equine arterivirus virus, lactate dehydrogenase-elevating viruses, porcine reproductive and respiratory syndrome viruses, or wobbly possum disease virus have a similar particle organization but lack proteins 2a′, 2b′, 3′, and 4′
None of these four arteriviruses were mentioned in the First Report (1971) of the International Committee on Nomenclature of Viruses (ICNV), the predecessor of the ICTV [58]. In the Second ICTV Report (1976), EAV and LDV were considered related due to their similar protein composition [59] and listed as unclassified members of the family Togaviridae because of the similarity of their virions to those of classified togaviruses [26]. SHFV joined this group of unclassified togaviruses in the Third (1979) and Fourth (1982) Reports [39, 40]. During subsequent years, the family Togaviridae was reorganized thoroughly. As a consequence, the genus Arterivirus was established and included in the family Togaviridae as a taxonomic home for EAV. LDV was considered a second member of this genus, and SHFV was removed from the family Togaviridae and placed into a group with hepatitis C virus in the newly established family Flaviviridae [51, 56]. However, the Fifth Report failed to take into account that EAV replication occurs via a 5′-3′-coterminal set of nested subgenomic mRNAs, a strategy not used by togaviruses or flaviviruses, but is characteristic of coronaviruses [20, 21, 28, 48]. Subsequent genomic studies confirmed the relatedness of EAV, LDV, PRRSV, and SHFV. Therefore, in the ICTV’s Sixth Report (1995), all four viruses were assigned to a free-floating genus Arterivirus [18]. The ICTV’s Seventh Report (2000) formalized the relationship of arteriviruses and coronaviruses by including the genus Arterivirus in a new family, Arteriviridae, and by including this family and the family Coronaviridae in a new order, Nidovirales. EAV, LDV, PRSSV, and SHFV were assigned as single members to the four arteriviral species Equine arteritis virus, Lactate dehydrogenase-elevating virus, Porcine respiratory and reproductive syndrome virus, and Simian hemorrhagic fever virus, respectively [14, 23]. This taxonomy was upheld in the ICTV’s Eight (2005) and Ninth Reports (2011) and is still valid at the time of writing [24, 46]. A summary of this arterivirus taxonomy is given in Table 1.
Current challenges for arterivirus taxonomy
Novel arteriviruses require classification
Eleven simian arteriviruses were discovered in recent years by next-generation sequencing in apparently healthy African cercopithecid nonhuman primates. Kibale red colobus viruses 1 and 2 (KRCV-1/2) were found in wild Ugandan red colobus (Procolobus [Piliocolobus] rufomitratus tephrosceles Elliot, 1907) from Uganda’s Kibale National Park [10, 35]. Kibale red-tailed guenon viruses 1 and 2 (KRTGV-1/2) were discovered in the same location in healthy red-tailed monkeys (Cercopithecus ascanius Audebert, 1799) [36]. Mikumi yellow baboon virus 1 (MYBV-1) was found in yellow baboons (Papio cynocephalus Linnaeus, 1766) in Tanzania’s Mikumi National Park, and the closely related Southwest baboon virus 1 (SWBV-1) was detected in healthy captive olive baboons (Papio anubis, Lesson 1827) at the Southwest National Primate Research Center in San Antonio, Texas, USA [9]. Results of a recent study identified two novel simian arteriviruses, Pebjah virus (PBJV) and simian hemorrhagic encephalitis virus (SHEV), as etiological agents of historical simian hemorrhagic fever outbreaks in Asian macaques, but their natural hosts remain unknown [37]. During yet unpublished studies, Drakensberg Mountain vervet virus 1 (DMVV-1) and Kafue kinda-chacma baboon virus 1 (KKCBV-1) were discovered in vervet monkeys (Chlorocebus pygerythrus Cuvier, 1821) and Kinda baboons (Papio kindae Lönnberg, 1919) in South Africa and Zambia, respectively. Finally, a novel simian arterivirus was found in a DeBrazza’s monkey (Cercopithecus neglectus Schlegel, 1876) in Cameroon and its sequence was deposited in GenBank (Ng et al., unpublished).
Analyses of the genomes of these eleven viruses clearly identify them as members of the family Arteriviridae. In fact, all arterivirus-defining elements (Figs. 1 and 2) can be identified unambiguously. Such elements include: open reading frame (ORF)1a and ORF1b encoding nonstructural proteins, ORFs2a–7 encoding structural proteins, and joined ORF1a and ORF1b (through a -1 programmed ribosomal frameshift site + RNA pseudoknot) encoding a polyprotein that is cleaved into main proteinase 3CLPRO, RNA-dependent RNA polymerase, putative zinc-binding domain (ZBD), helicase (HEL1), and uridylate-specific endoribonuclease (NendoU) [24, 47]) (Fig. 2). More importantly, all eleven simian arteriviruses are characterized by the presence of additional ORFs that are characteristic of the SHFV genome, but which are absent from the genomes of all other currently characterized arteriviruses (Fig. 2).

Organization of arterivirus genomes. Individual arterivirus open reading frames (ORFs) drawn to scale are shown in colors. ORF1a (green) encodes polyprotein 1a (pp1a). ORF1a and ORF1b (yellow) can be joined through a “-1” programmed ribosomal frameshift to express polyprotein 1ab. pp1a and pp1ab are translated directly from genomic RNA and are subsequently proteolytically cleaved into numerous nonstructural proteins, many of which are part of the replicase complex. Nonstructural protein 2 transframe (nsp2TF) is produced through a “-2” programmed ribosomal frameshift by most, but not all, arteriviruses. The remaining ORFs are translated from subgenomic RNAs. Purple ORFs (four shades) encode minor structural proteins (envelope protein E, glycoproteins 2b, 3 and 4); blue ORFs (three shades) encode the major structural proteins GP5 (glycoprotein 5), M (matrix protein), and N (nucleocapsid protein). Red ORFs (four shades), present only in simian arterivirus (simartevirus) genomes, encode minor structural proteins of unknown function (see also Fig. 1B). Virus genomes are labeled by retained or new virus abbreviations with previous designations in grey in parentheses. Simian arteriviruses (simarteviruses) are: KRTGV-1/2, Kibale red-tailed guenon viruses 1 and 2; DeBMV-1, DeBrazza’s monkey virus 1; PBJV, Pebjah virus; SHFV, simian hemorrhagic fever virus; KKCBV-1, Kafue kinda-chacma baboon virus 1; MYBV-1, Mikumi yellow baboon virus 1; SWBV-1, Southwest baboon virus 1; KRCV-1/2, Kibale red colobus viruses 1 and 2; SHEV, simian hemorrhagic encephalitis virus; DMVV-1; Drakensberg Mountain vervet virus 1. Murine and porcine arteriviruses (rodarteviruses) are: LaDV-1/2, lactate dehydrogenase-elevating viruses 1 and 2; PRRSV-1/2, and porcine reproductive and respiratory syndrome viruses 1 and 2. Nesomyid arteriviruses (nesarteviruses) contain: APRAV-1, African pouched rat virus 1. Equine arteriviruses (equarteviruses) contain: EAV, equine arteritis virus. Possum arterviruses (diparteviruses) include: WPDV, wobbly possum disease virus. For GenBank accession numbers used for the analyses, see Table 2 (color figure online)
A novel arterivirus was recently discovered in a forest giant pouched rat (Cricetomys emini Wroughton, 1910) in Cameroon (GenBank and Ng et al., unpublished). In 2012, Dunowska et al. published the partial genome of a novel arterivirus-like pathogen, wobbly possum disease virus (WPDV), found in common brushtails (Diprodontia: Phalangeridae: Trichosurus vulpecula Kerr, 1972), a marsupial introduced to New Zealand [22]. The complete genomic sequences of WPDV were deposited in GenBank [unpublished data], and the virus was isolated in tissue culture in 2015 [27]. Both the virus from the forest giant pouched rat and WPDV genomes contain all arterivirus-defining elements described above, and recent electron-microscopic studies revealed that the WPDV particles assume the typical arterivirion shape (spherical to icosahedral particles) [22, 27]. However, the WPDV genome sequence is highly divergent from the genomes of classified arteriviruses [22, 47], and WPDV is the first arterivirus found in a non-eutherian host.
The current arterivirus nomenclature is discouraged by the International Code of Virus Classification and Nomenclature
The ICVCN has several Rules to discourage naming of taxa in ambiguous ways to avoid confusion, such as:
-
“The essential principles of virus nomenclature are … to avoid or reject the use of names which might cause error or confusion” (Rule 2.1(ii)) [4, 30]
-
“New names shall not duplicate approved names. New names shall be chosen such that they are not closely similar to names that are in use currently or have been in use in the recent past” (Rule 3.14) [4, 30]
-
“A species name shall consist of as few words as practicable but be distinct from names of other taxa” (Rule 3.21) [4, 30]
-
“A species name must provide an appropriately unambiguous identification of the species” (Rule 3.22) [4, 30]
The current arterivirus nomenclature has species and virus names that are identical in spelling and are only differentiated by the presence or absence of italics and first word capitalization (e.g., the single member of the species Equine arteritis virus is called equine arteritis virus). Consequently, species names and virus names are often confused in writing, and virus abbreviations are often wrongly assigned to species rather than viruses. This practice obfuscates the distinction between species, which are conceptual entities, and viruses, which are physical entities [16, 31, 53].
Another problem associated with the current arterivirus nomenclature is that the current family and genus names are only differentiated by their suffixes (“-viridae” vs. “-virus”) but have an identical word stem (“Arteri-“). Since species-specific suffixes for the members of individual taxon hierarchies [55] are not yet accepted in the virology community, the members of both the family Arteriviridae and the genus Arterivirus are currently called simply “arteriviruses.” The word “arterivirus” will therefore become ambiguous as soon as a second genus is established in the family, as it will not be clear then whether “arterivirus” refers to the members of the one genus or to the members of the entire family.
Finally, the current species names Lactate dehydrogenase-elevating virus, Porcine respiratory and reproductive syndrome virus, and Simian hemorrhagic fever virus contradict Rule 3.21 (“…shall consist of as few words as practicable”).
New classification and nomenclature
The currently approved arterivirus classification relies on three key characteristics [24]:
-
genomic organization and size. The genomic organization considered typical of arteriviruses is based on the genome organization of EAV (Fig. 2). EAV, LDV, and PRRSV genomes are 12.7 to 15.4 kb in length. The replicase ORFs 1a and 1b encoding replicase precursor polyproteins pp1a/pp1ab are followed by genes encoding envelope (E) protein, glycoproteins (GP2b-GP5), membrane (M) protein, and nucleocapsid protein (N) (5′-1a/1b-2a-2b-3-4-5a-5-6-7-3′), which justifies their grouping (Fig. 2). The SHFV genome contains the same genomic architecture but also possesses 4 additional ORFs immediately downstream of ORF1b (2a′, 2b′, 3′, 4′). The SHFV genome is ≈15.7 kb in length;
-
monophyly. Phylogenetic analyses clearly separate EAV, LDV, PSSRV, and SHFV into four clades. These analyses are based on the similarity of the ORF1b nucleotide sequences or the encoded pp1b amino acid sequences and/or on homologous and alignable portions of coding-complete viral genomes (thus excluding the additional 4 SHFV ORFs) [36, 47]; and
-
host range. EAV, LDV, PRRSV, and SHFV are known to infect equids, rodents, suids, and nonhuman primates, respectively.
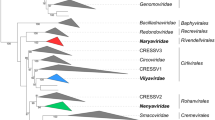
The criteria listed above indicate that DMVV-1, KKCBV-1, KRCV-1, KRCV-2, KRTGV-1, KRTGV-2, MYBV-1, PBJV, SHEV, SWBV-1, and the viruses from DeBrazza’s and vervet monkeys should be grouped together with SHFV because these viruses 1) share the typical SHFV genomic architecture containing the SHFV-specific additional reading frames 2a′, 2b′, 3′, and 4′ (Fig. 2); 2) are more closely related to each other in pairwise distance analyses of genome-encoded proteins than to the proteins of the remaining arteriviruses (Fig. 3); 3) cluster more closely with SHFV in ORF1b phylogenetic analysis than with any of the other known arteriviruses (Fig. 4); and 4) infect cercopithecids (Old World monkeys), whereas none of the other arteriviruses are known to infect nonhuman primates. However, the pairwise genetic distances of the twelve simian arteriviruses are less than 50 % identical to each other on the nucleotide level. Such genetic distances are similar to the distance observed between the genomes of PRRSVs and LDV, PRSSVs and EAV, or LDV and EAV, all of which are currently classified members of separate species. These data indicate that eleven of the newly discovered simian arteriviruses might be representatives of novel arterivirus species, rather than novel members of the current species Simian hemorrhagic fever virus (Fig. 3).

Pairwise sequence comparison of the arterivirus proteomes. Coding regions from each arterivirus genome (Fig. 2) were aligned in CLC Genomics Workbench version 6, and a pairwise comparison matrix was constructed. Only orthologous genes common to all known arteriviruses were used (i.e., simian arterivirus-/simartevirus-specific ORFs were ignored). Numbers represent the percent nucleotide identity between two viruses, with red highlighting virus pairs with relatively high degrees of similarity and white showing virus pairs with lower similarity. Virus abbreviations and sequences sources for the analyses are identical to those outlined in Fig. 2 and Table 2

Phylogenetic relationships among classified and unclassified arteriviruses based on ORF1b amino acid sequences (see Fig. 2 for genomes and virus abbreviations). Silhouettes represent mammals in which each virus was found, with question marks indicating uncertain natural hosts (viruses discovered in captive macaques). Sequences were compiled from Table 2. ORF1b nucleotide sequences were aligned using a codon-guided version of the multiple alignment using fast fourier transform (MAFFT) method [29] implemented in the computer program TranslatorX [1]. Poorly aligned sites were removed using the Gblocks alignment cleaning method [17] and the resulting sequence was translated into amino acid sequences. Phylogenetic analyses were then conducted on aligned sequences (final alignment length of 1,055 positions) using the Neighbor-Joining method [45] with the Poisson distance correction method [60]. Robustness of phylogenetic groupings was assessed using 1,000 bootstrap replicates of the data [25]; only bootstrap values >50 % are shown. The scale bar represents amino acid substitutions per site. Analyses were conducted in MEGA7. The topology of the tree shown is identical to that of trees constructed using maximum likelihood analyses of nucleic acid sequences (not shown)
To confirm these results and to achieve an objective resolution of arterivirus relationships, we used the PAirwise Sequence Comparison (PASC) methodology [11] and tools available from the National Center for Biotechnology Information (NCBI). The PASC analysis aligns nucleotides in conserved protein or nucleotide regions of each virus pair. The resulting histograms visualize the distribution of the number of virus pairs at each identity percentage. A typical histogram has peaks that represent different taxon levels. The percentages of the lowest points of the valleys between the peaks can be used as taxon demarcation criteria. The results of the arterivirus PASC analysis are shown in Figure 5 (updates can be accessed at the NCBI website at http://www.ncbi.nlm.nih.gov/sutils/pasc/viridty.cgi?textpage=overview). Results from PASC indicate that the most appropriate genus cut-off for arteriviruses is 39–41 %. The most appropriate species cut-off would be 71–77 %, which is close to species cutoffs set for other viral families. These cut-offs result in a split of the current genus Arterivirus into 5 genera, a split of the current species Porcine respiratory and reproductive syndrome virus into two species (one for “European/Lelystad” PRRSV type 1 and one for “North American” PRRSV type 2), and the split of the species Simian hemorrhagic fever virus into ten species.

PAirwise Sequence Comparison (PASC) analysis using a modified basic local alignment search tool (BLAST) algorithm [11]. The resulting histograms, visualizing the distribution of the number of arterivirus pairs at each identity percentage, confirm results shown in Figure 3. The x-axis shows percent identity (0–100 %) and the y-axis shows the number of compared arterivirus sequence pairs. A) original color coding assigned by the NCBI PASC tool, showing disarray. B) reassigned color coding after implementation of the arterivirus taxonomy proposed herein (color figure online)
To accommodate these results, the taxonomy outlined in Table 2 is proposed. Accordingly, the current genus Arterivirus is replaced with five genera: Dipartevirus (sigil of the mammalian order Diprotodontia + arterivirus) for WPDV; Equartevirus (sigil of the mammalian family Equidae + arterivirus) for EAV; Nesartevirus (sigil of the mammalian family Nesomyidae + arterivirus) for the forest giant pouched rat virus; Rodartevirus (sigil of the mammalian families Rodentia and Artiodactyla + arterivirus) for LDV and PRRSV; and Simartevirus (sigil of the mammalian infraorder Simiiformes + arterivirus) for all simian arteriviruses. These genus names were consciously selected to
-
not contain numbers or references to Greek letters or Arabic numbers (e.g., “Alphaarterivirus, Betaarterivirus” or “Arterivirus 1,” Arterivirus 2”) for ease of memorization and use in vernacular writing and oral communication. Future establishment of new genera in the family will not be hampered by the length of the Greek alphabet or by the addition of large numbers. In addition, by not choosing such names, the visually unpleasing double vowels aa (Alph aa rterivirus, Bet aa rterivirus etc.), which formally would require diareses, are avoided;
-
not contain the complete word stems of hosts or geographic locations. This way, the genus names should remain uncontroversial even if members of the genus are discovered in areas or hosts divergent from the founding members;
-
contain mnemonics for easier memorization. For instance, the genus name Equ artevirus will be easier to remember for equine arteritis virus than, for instance, Alphaarterivirus or Arterivirus 1. But as the three letters “Equ” do not convey the same meaning as “equine” to the casual reader, additional genus members, not stemming from equids, can be included without making the genus name misleading;
-
not contain complete word stems of higher taxa (e.g., “arteri” or “nido”) but to again use contractions (“arte”). This choice will allow adherence to the ICVCN “stability rule”: i.e., the chosen genus names could be retained even if future taxonomic reorganizations would require the dissolution of the family Arteriviridae or the order Nidovirales, or if individual genera would have to be moved to separate families.
To differentiate species and virus names clearly, the ICTV-supported proposal to employ non-Latinized binomial species names was followed [54]. In the proposed classification, the virus name “equine arteritis virus” and its abbreviation “EAV” are retained, therefore avoiding any significant impact of this taxonomic proposal on the arterivirus research community. However, the name of the species for EAV, which rarely has to be used in daily proceedings by virologists, is changed to Equid equartevirus. Similar changes are introduced for species of the remaining genera.
The virus name “lactate dehydrogenase-elevating virus” is retained, but its current abbreviation (LDV) is changed to LaDV to differentiate it from lymphocystis disease virus (LDV, an iridovirus of teleost fishes). To reflect the relatively large distance between the two “strains” LDV-P and LDV-C, both strains are elevated to virus status (LaDV-1 and LaDV-2). The species name harboring these viruses is changed to Muroid rodartevirus. The current species Porcine respiratory and reproductive syndrome virus is split into Suid 1 rodartevirus and Suid 2 rodartevirus for “European/Lelystad” porcine respiratory and reproductive syndrome virus type 1 (now renamed PRRSV-1), and “North American” PRRSV type 2 (now renamed PRRSV-2), respectively.
The genus Simartevirus includes ten species. Seven of these species are named after the species for the hosts the viruses were discovered in: Cercopithecus 1 simartevirus for KRTGV-1 and KRTGV-2; Cercopithecus 2 simartevirus for the arterivirus from the DeBrazza’s monkey; Chlorocebus 1 simartevirus for DMVV-1; Papio 1 simartevirus for MYBV-1 and SWBV-1; Papio 2 simartevirus for KKCBV-1; Procolobus 1 simartevirus for KRCV-1; Procolobus 2 simartevirus for KRCV-2. The hosts of the three simarteviruses known to have caused epizootics among captive macaques (PBJV, SHEV, SHFV) are unknown. Consequently, the names of the species these viruses belong to were chosen based on the cities in which the outbreaks occurred: Alamogordo simartevirus for PBJV; Bethesda simartevirus for SHFV; and Sukhumi simartevirus for SHEV. Host names were once again used for the species established for WPDV (Trichosurus 1 dipartevirus) and the arterivirus from the forest giant pouched rat (Cricetomys 1 nesartevirus). The virus found in DeBrazza vervet monkeys was named DeBrazza’s monkey virus 1 (DeBMV-1), and the virus found in forest giant pouched rats was named African pouched rat virus 1 (APRAV-1). Pronunciation guidelines for all arterivirus names are listed in Table 3.
Together, this new taxonomy resolves all previous disagreements with the ICVCN while at the same time accommodating all newly discovered arteriviruses and providing relative consistency with previously established virus names and abbreviations.
Proposal for nomenclature below the species level
To improve retrieval of arterivirus genomic or protein data from electronic databases, we propose that arterivirus strains/variants/isolates be named according to the NCBI-supported naming scheme recently proposed for filoviruses [32–34] and rotaviruses [41]: <virus name>/<strain>/<isolation host-suffix>/<country of sampling>/<year of sampling>/<genetic variant designation>-<isolate designation>. References [32–34] contain detailed instructions on how to fill in the individual fields of this naming template in a standardized manner. We also strongly suggest not using the word “strain” indiscriminately [32–34]. A consortium of arterivirus experts should be established under the auspices of the ICTV Arteriviridae Study Group to decide on the precise strain/variant/isolate names for the several thousand arteriviruses currently indexed in databases such as GenBank.
Proposal for adjustment of gene and protein designations
NCBI is currently developing gene and protein naming standards for viruses to improve electronic database management. Generally, we propose not to change gene and protein names at this point.
Future prospects
The taxonomy of the family Arteriviridae will most likely have to be amended and expanded repeatedly in the future. Evidence is accumulating that the diversity of arteriviruses is higher than currently appreciated [43, 49, 50]. Furthermore, the paraphyly of horses, nonhuman primates, pigs, possums, and rodents within the class Mammalia [42] suggests that many other mammalian orders may host as yet undiscovered arteriviruses. The establishment of methods-of-choice for arterivirus taxon demarcation and the implementation of unambiguous taxon and virus names in line with ICVCN will prove to be a solid foundation for the taxonomic accommodation of arteriviruses that will undoubtedly be discovered in the future.
References
Abascal F, Zardoya R, Telford MJ (2010) TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res 38:W7–13
Adams MJ, Carstens EB (2012) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch Virol 157:1411–1422
Adams MJ, King AM, Carstens EB (2013) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch Virol 158:2023–2030
Adams MJ, Lefkowitz EJ, King AM, Carstens EB (2013) Recently agreed changes to the International Code of Virus Classification and Nomenclature. Arch Virol 158:2633–2639
Adams MJ, Carstens EB (2014) Changes to the Statutes and Subcommittees of the International Committee on Taxonomy of Viruses (2014). Arch Virol 159:2829–2830
Adams MJ, Lefkowitz EJ, King AM, Carstens EB (2014) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2014). Arch Virol 159:2831–2841
Adams MJ, Lefkowitz EJ, King AM, Bamford DH, Breitbart M, Davison AJ, Ghabrial SA, Gorbalenya AE, Knowles NJ, Krell P, Lavigne R, Prangishvili D, Sanfaçon H, Siddell SG, Simmonds P, Carstens EB (2015) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2015). Arch Virol 160:1837–1850
Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Ali Khan S, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JA, Daszak P, Lipkin WI (2013) A strategy to estimate unknown viral diversity in mammals. MBio 4:e00598–00513
Bailey AL, Lauck M, Sibley SD, Pecotte J, Rice K, Weny G, Tumukunde A, Hyeroba D, Greene J, Correll M, Gleicher M, Friedrich TC, Jahrling PB, Kuhn JH, Goldberg TL, Rogers J, O’Connor DH (2014) Two novel simian arteriviruses in captive and wild baboons (Papio spp.). J Virol 88:13231–13239
Bailey AL, Lauck M, Weiler A, Sibley SD, Dinis JM, Bergman Z, Nelson CW, Correll M, Gleicher M, Hyeroba D, Tumukunde A, Weny G, Chapman C, Kuhn JH, Hughes AL, Friedrich TC, Goldberg TL, O’Connor DH (2014) High genetic diversity and adaptive potential of two simian hemorrhagic fever viruses in a wild primate population. PLoS One 9:e90714
Bao Y, Chetvernin V, Tatusova T (2014) Improvements to pairwise sequence comparison (PASC): a genome-based web tool for virus classification. Arch Virol 159:3293–3304
Benfield DA, Nelson E, Collins JE, Harris L, Goyal SM, Robison D, Christianson WT, Morrison RB, Gorcyca D, Chladek D (1992) Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J Vet Diagn Invest 4:127–133
Breitbart M, Rohwer F (2005) Here a virus, there a virus, everywhere the same virus? Trends Microbiol 13:278–284
Brinton MA, Godeny EK, Horzinek MC, Meulenberg JJM, Murtaugh MP, Plagemann PG, Snijder EJ (2000) Family Arteriviridae. In: van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB (eds) Virus taxonomy—seventh report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, pp 851–857
Bryans JT, Crowe ME, Doll ER, McCollum WH (1957) Isolation of a filterable agent causing arteritis of horses and abortion by mares; its differentiation from the equine abortion (influenza) virus. Cornell Vet 47:3–41
Calisher CH, Mahy BW (2003) Taxonomy: get it right or leave it alone. Am J Trop Med Hyg 68:505–506
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Cavanagh D, Brain DA, Brinton MA, Enjuanes L, Holmes KV, Horzinek MC, Lai MMC, Laude H, Plagemann PGW, Siddell SG, Spaan WJM, Taguchi F, Talbot PJ (1995) Genus Arterivirus. In: Murphy FA, Fauquet CM, Bishop DHL, Ghabrial SA, Jarvis AW, Martelli GP, Mayo MA, Summers MD (eds) Virus taxonomy—sixth report of the International Committee on Taxonomy of Viruses. Springer, Vienna, pp 412–414
Collins JE, Benfield DA, Christianson WT, Harris L, Hennings JC, Shaw DP, Goyal SM, McCullough S, Morrison RB, Joo HS et al (1992) Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Invest 4:117–126
de Vries AA, Chirnside ED, Bredenbeek PJ, Gravestein LA, Horzinek MC, Spaan WJ (1990) All subgenomic mRNAs of equine arteritis virus contain a common leader sequence. Nucleic Acids Res 18:3241–3247
den Boon JA, Snijder EJ, Chirnside ED, de Vries AA, Horzinek MC, Spaan WJ (1991) Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J Virol 65:2910–2920
Dunowska M, Biggs PJ, Zheng T, Perrott MR (2012) Identification of a novel nidovirus associated with a neurological disease of the Australian brushtail possum (Trichosurus vulpecula). Vet Microbiol 156:418–424
Enjuanes L, Spaan W, Snijder E, Cavanagh D (2000) Order Nidovirales. In: van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB (eds) Virus taxonomy—seventh report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, California, USA, pp 827–834
Faaberg KS, Balasuriya UB, Brinton MA, Gorbalenya AE, Leung FC-C, Nauwynck H, Snijder EJ, Stadejek T, Yang H, Yoo D (2011) Family Arteriviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy—ninth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, London, pp 796–805
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fenner F (1976) Classification and nomenclature of viruses. Second report of the International Committee on Taxonomy of Viruses. Intervirology 7:1–115
Giles JC, Perrott MR, Dunowska M (2015) Primary possum macrophage cultures support the growth of a nidovirus associated with wobbly possum disease. J Virol Methods 222:66–71
Godeny EK, de Vries AA, Wang XC, Smith SL, de Groot RJ (1998) Identification of the leader-body junctions for the viral subgenomic mRNAs and organization of the simian hemorrhagic fever virus genome: evidence for gene duplication during arterivirus evolution. J Virol 72:862–867
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2011) The International Code of Virus Classification and Nomenclature. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy—ninth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, London, pp 1273–1277
Kuhn JH, Jahrling PB (2010) Clarification and guidance on the proper usage of virus and virus species names. Arch Virol 155:445–453
Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brister JR, Bukreyev AA, Caì Y, Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Gonzalez JP, Formenty P, Freiberg AN, Hensley LE, Honko AN, Ignatyev GM, Jahrling PB, Johnson KM, Klenk HD, Kobinger G, Lackemeyer MG, Leroy EM, Lever MS, Lofts LL, Mühlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Ryabchikova EI, Saphire EO, Shestopalov AM, Smither SJ, Sullivan NJ, Swanepoel R, Takada A, Towner JS, van der Groen G, Volchkov VE, Wahl-Jensen V, Warren TK, Warfield KL, Weidmann M, Nichol ST (2013) Virus nomenclature below the species level: a standardized nomenclature for laboratory animal-adapted strains and variants of viruses assigned to the family Filoviridae. Arch Virol 158:1425–1432
Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brister JR, Bukreyev AA, Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Hensley LE, Honko AN, Jahrling PB, Johnson KM, Kobinger G, Leroy EM, Lever MS, Mühlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Saphire EO, Smither SJ, Swanepoel R, Towner JS, van der Groen G, Volchkov VE, Wahl-Jensen V, Warren TK, Weidmann M, Nichol ST (2013) Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch Virol 158:301–311
Kuhn JH, Bào Y, Bavari S, Becker S, Bradfute S, Brauburger K, Rodney Brister J, Bukreyev AA, Caì Y, Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Gonzalez JP, Formenty P, Freiberg AN, Hensley LE, Hoenen T, Honko AN, Ignatyev GM, Jahrling PB, Johnson KM, Klenk HD, Kobinger G, Lackemeyer MG, Leroy EM, Lever MS, Mühlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Ryabchikova EI, Saphire EO, Shestopalov AM, Smither SJ, Sullivan NJ, Swanepoel R, Takada A, Towner JS, van der Groen G, Volchkov VE, Volchkova VA, Wahl-Jensen V, Warren TK, Warfield KL, Weidmann M, Nichol ST (2014) Virus nomenclature below the species level: a standardized nomenclature for filovirus strains and variants rescued from cDNA. Arch Virol 159:1229–1237
Lauck M, Hyeroba D, Tumukunde A, Weny G, Lank SM, Chapman CA, O’Connor DH, Friedrich TC, Goldberg TL (2011) Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS One 6:e19056
Lauck M, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Chapman CA, Ting N, Switzer WM, Kuhn JH, Friedrich TC, O’Connor DH, Goldberg TL (2013) Exceptional simian hemorrhagic fever virus diversity in a wild African primate community. J Virol 87:688–691
Lauck M, Alkhovsky SV, Bao Y, Bailey AL, Shevtsova ZV, Shchetinin AM, Vishnevskaya TV, Lackemeyer MG, Postnikova E, Mazur S, Wada J, Radoshitzky SR, Friedrich TC, Lapin BA, Deriabin PG, Jahrling PB, Goldberg TL, O’Connor DH, Kuhn JH (2015) Historical outbreaks of simian hemorrhagic fever in captive macaques were caused by distinct arteriviruses. J Virol 89:8082–8087
Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, Qin XC, Xu J, Holmes EC, Zhang YZ (2015) Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 4:e05378
Matthews RE (1979) Third report of the International Committee on Taxonomy of Viruses. Classification and nomenclature of viruses. Intervirology 12:129–296
Matthews RE (1982) Classification and nomenclature of viruses. Fourth report of the International Committee on Taxonomy of Viruses. Intervirology 17:1–199
Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Banyai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gomara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreno V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M (2011) Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol 156:1397–1413
Meredith RW, Janecka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simao TL, Stadler T, Rabosky DL, Honeycutt RL, Flynn JJ, Ingram CM, Steiner C, Williams TL, Robinson TJ, Burk-Herrick A, Westerman M, Ayoub NA, Springer MS, Murphy WJ (2011) Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334:521–524
Mittelholzer C, Stadejek T, Johansson I, Baule C, Ciabatti I, Hannant D, Paton D, Autorino GL, Nowotny N, Belak S (2006) Extended phylogeny of equine arteritis virus: division into new subgroups. J Vet Med B Infect Dis Vet Public Health 53:55–58
Notkins AL, Scheele C (1963) An infectious nucleic acid from the lactic dehydrogenase agent. Virology 20:640–642
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Snijder EJ, Brinton MA, Faaberg KS, Godeny EK, Gorbalenya AE, MacLachlan NJ, Mengeling WL, Plagemann PGW (2005) Family Arteriviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds) Virus taxonomy—eighth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, San Diego, pp 965–974
Snijder EJ, Kikkert M, Fang Y (2013) Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163
Spaan WJM, den Boon JA, Bredenbeek PJ, Chirnside ED, Noten AFH, Snijder EJ, de Vries AAF, Horzinek MC (1990) Comparative and evolutionary aspects of Coronaviral, Arteriviral, and Toroviral genome structure and expression. In: Brinton MA, Heinz FX (eds) New aspects of positive-strand RNA viruses. American Society for Microbiology, Washington, DC, pp 12–19
Stadejek T, Stankevicius A, Storgaard T, Oleksiewicz MB, Belak S, Drew TW, Pejsak Z (2002) Identification of radically different variants of porcine reproductive and respiratory syndrome virus in Eastern Europe: towards a common ancestor for European and American viruses. J Gen Virol 83:1861–1873
Stadejek T, Oleksiewicz MB, Potapchuk D, Podgorska K (2006) Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in eastern Europe support the definition of new genetic subtypes. J Gen Virol 87:1835–1841
Strauss JH (1991) Family Togaviridae. In: Francki RIB, Fauquet CM, Knudson DL, Brown F (eds) Classification and nomenclature of viruses—fifth report of the International Committee on Taxonomy of Viruses. Springer, Vienna, pp 216–222
Tauraso NM, Shelokov A, Palmer AE, Allen AM (1968) Simian hemorrhagic fever. 3. Isolation and characterization of a viral agent. Am J Trop Med Hyg 17:422–431
Van Regenmortel MH (2007) Virus species and virus identification: past and current controversies. Infect Genet Evol 7:133–144
Van Regenmortel MH, Burke DS, Calisher CH, Dietzgen RG, Fauquet CM, Ghabrial SA, Jahrling PB, Johnson KM, Holbrook MR, Horzinek MC, Keil GM, Kuhn JH, Mahy BW, Martelli GP, Pringle C, Rybicki EP, Skern T, Tesh RB, Wahl-Jensen V, Walker PJ, Weaver SC (2010) A proposal to change existing virus species names to non-Latinized binomials. Arch Virol 155:1909–1919
Vetten HJ, Haenni AL (2006) Taxon-specific suffixes for vernacular names. Arch Virol 151:1249–1250
Wengler G (1991) Family Flaviviridae. In: Francki RIB, Fauquet CM, Knudson DL, Brown F (eds) Classification and nomenclature of viruses—fifth report of the International Committee on Taxonomy of Viruses. Springer, Vienna, pp 223–233
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F et al (1991) Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Wildy P (1971) Classification and nomenclature of viruses. First report of the International Committee on Nomenclature of Viruses. Karger, Basel
Zeegers JJ, Van der Zeijst BA, Horzinek MC (1976) The structural proteins of equine arteritis virus. Virology 73:200–205
Zuckerkandl E, Pauling L (1965) Evolutionary divergence and convergence in proteins. In: Bryson V, Vogel HJ (eds) Evolving genes and proteins. Academic Press, New York, pp 97–166
Acknowledgments
This work was funded in part through Battelle Memorial Institute’s prime contract with the US National Institute of Allergy and Infectious Diseases (NIAID) under Contract No. HHSN272200700016I. Battelle employees involved in this work are: L.B. and J.W. Subcontractors to Battelle Memorial Institute who performed this work are: J.H.K and E.P., employees of Tunnell Government Services, Inc.; A.N.C. the owner of Logos Consulting, Inc.; and M.G.L., an employee of Lovelace Respiratory Research Institute. This work was also funded in part through National Institutes of Health (NIH) grant TW009237 as part of the joint NIH-NSF Ecology of Infectious Disease program, grant R01 AI077376, and by the Office of Research Infrastructure Programs (ORIP) grant P51OD011106. This research was also supported in part by the Intramural Research Program of the National Institutes of Health, National Library of Medicine (Y.B.), and benefited from contributions from the PREDICT project of the United States Agency for International Development (USAID) Emerging Pandemic Threats Program. We thank the Cameroon government for their support during the implementation of this project.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kuhn, J.H., Lauck, M., Bailey, A.L. et al. Reorganization and expansion of the nidoviral family Arteriviridae . Arch Virol 161, 755–768 (2016). https://doi.org/10.1007/s00705-015-2672-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2672-z