
Abstract
A set of proposals to rationalize and extend the taxonomy of the family Parvoviridae is currently under review by the International Committee on Taxonomy of Viruses (ICTV). Viruses in this family infect a wide range of hosts, as reflected by the longstanding division into two subfamilies: the Parvovirinae, which contains viruses that infect vertebrate hosts, and the Densovirinae, encompassing viruses that infect arthropod hosts. Using a modified definition for classification into the family that no longer demands isolation as long as the biological context is strong, but does require a near-complete DNA sequence, 134 new viruses and virus variants were identified. The proposals introduce new species and genera into both subfamilies, resolve one misclassified species, and improve taxonomic clarity by employing a series of systematic changes. These include identifying a precise level of sequence similarity required for viruses to belong to the same genus and decreasing the level of sequence similarity required for viruses to belong to the same species. These steps will facilitate recognition of the major phylogenetic branches within genera and eliminate the confusion caused by the near-identity of species and viruses. Changes to taxon nomenclature will establish numbered, non-Latinized binomial names for species, indicating genus affiliation and host range rather than recapitulating virus names. Also, affixes will be included in the names of genera to clarify subfamily affiliation and reduce the ambiguity that results from the vernacular use of “parvovirus” and “densovirus” to denote multiple taxon levels.
Similar content being viewed by others


Introduction
A set of proposals to update the taxonomy of the family Parvoviridae has been submitted by a review group that includes all members of the International Committee on Taxonomy of Viruses (ICTV) Parvoviridae Study Group (SG), and is currently under review. Until a final ICTV decision is reached, the proposal can be downloaded at http://talk.ictvonline.org/files/proposals/taxonomy_proposals_vertebrate1/default.aspx. The taxonomy of this family was last modified in 2004, prior to publication of the 8th ICTV Report [13], and is now significantly dated. In the interim, many new candidate viruses and previously unsuspected viral hierarchies have been identified, often by the use of viral discovery approaches that rely on polymerase chain reaction DNA amplification. Unfortunately, this approach typically confounds characterization of complex secondary structures in the viral hairpin telomeres that are essential for viability [8, 10], making the recovery of viruses from DNA challenging. To accommodate these important new viruses, while avoiding inclusion of viral sequence fragments integrated into host genomes [1, 9] or metagenomic data that lack integrity or clear host attribution (for example, a full-length Blatella germanica densovirus-like virus sequence, GenBank JQ320376, with a probable cockroach host that was identified in bat faeces [7]), the SG developed a polythetic definition of a virus in the family Parvoviridae. This requires the complete DNA sequence of all viral protein-coding sequences but no longer absolutely requires isolation of a viable virus provided an infectious etiology is supported by the structure and arrangement of the genome, serology, or other biological data. The viral definition used throughout these proposals is: “In order for an agent to be classified in the family Parvoviridae, it must be judged to be an authentic parvovirus on the basis of having been isolated and sequenced or, failing this, on the basis of having been sequenced in tissues, secretions, or excretions of unambiguous host origin, supported by evidence of its distribution in multiple individual hosts in a pattern that is compatible with dissemination by infection. The sequence must be in one piece, contain all the non-structural (NS) and viral particle (VP) coding regions, and meet the size constraints and motif patterns characteristic of the family”.
This definition allows inclusion of 134 new viruses and virus strains in the family Parvoviridae, together with 47 of the 53 previously recognized isolates [14]. Six recognized viruses for which no sequence information is currently available have been withdrawn from the formal taxonomy pending further analysis but will remain listed in subsequent reports as unassigned in their current genus. To improve taxonomic clarity and to facilitate the ready assimilation of present and future candidate viruses, a root-and-branch re-evaluation of the taxonomic structure and nomenclature of the family was also instituted, leading to the development of new systematic guidelines. Proposed changes to the taxonomy are summarized in Table 1. In the subfamily Parvovirinae, these changes include the introduction of three new genera and the expansion of five existing genus names with the affixes “parvo” or “proto”. In the subfamily Densovirinae, proposed changes include the introduction of two new genera for shrimp viruses and the expansion of the existing genus names Iteravirus and Densovirus to Iteradensovirus and Ambidensovirus, respectively. In both subfamilies, species identity levels will be lowered, numbered, binomial species names adopted, and new species introduced.
Changes in taxonomic structure and nomenclature
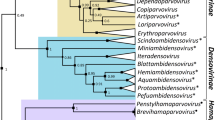
Taxon demarcation criteria
Parvoviruses encode two gene cassettes: an NS gene essential for replication and a VP gene encoding various forms of the structural protein [reviewed in 2, 14]. The amino acid sequence of the NS1 protein is used for phylogenetic analysis in the current proposals. NS1 is a multidomain ~70-80 kDa nuclear phosphoprotein that encodes highly conserved enzymatic activities, including a site-specific DNA-binding and single-strand endonuclease function and an AAA+ helicase [reviewed in 2, 3]. These well-conserved domains facilitate amino acid sequence alignment, allowing insights from structural biology and the derivation of a reliable sequence-based phylogeny (see the summary tree in Fig. 1). However, the core capsid protein sequence (defined as the smallest VP protein that contains all residues comprising the virion shell, as determined by X-ray crystallography) was analyzed in parallel, with conspicuously similar overall results although the data appeared less reliable at the greater distances apparent between the two subfamilies. Thus, the proposed taxonomic changes are supported by protein alignments of both major viral proteins. Previously, genera were defined largely by non-quantifiable criteria, including helper-virus requirements and genome characteristics, which provided little taxonomic structure [13, 14]. The current proposals add the requirement that all viruses in a genus should be monophyletic and encode NS1 proteins that are generally >30 % identical to each other at the amino acid sequence level but <30 % identical to those of other genera as determined by pairwise sequence alignments. Within the subfamily Parvovirinae, these criteria work well to separate all current and proposed genera, with the minor exception of the proposed genus Erythroparvovirus, where marginally greater divergence is evident between some virus pairs. The resulting eight genera in this subfamily are well supported by phylogenic analysis, as illustrated in Fig. 1 and detailed in the proposals.

Phylogenetic tree showing genera in the family Parvoviridae. Phylogenetic analysis based on the amino acid sequence of the viral replication initiator protein, NS1, which contains a conserved AAA+ helicase domain corresponding to the Parvo_NS1 Pfam domain: http://pfam.sanger.ac.uk/family/Parvo_NS1. This region was aligned by incorporating insights from structural biology using the ehmmalign application in EMBASSY [6], and sequences flanking the Pfam domain were aligned using the modification of the Needleman-Wunsch local alignment method [11] as implented in MOE-Align (http://www.chemcomp.com). Pairwise p-distance matrices were constructed from this alignment using MEGA version 5.10 [12]. Bayesian trees were calculated over one billion iterations using BEAST [5], using a Yule model of speciation and an exponential relaxed molecular clock [4]. Trees were viewed in FigTree (part of BEAST) in ultrametric format on an arbitrary scale, midpoint-rooted, and with posterior probability scores indicated at statistically significant nodes. Bold type in genus names indicates affixes used to expand existing names. Asterisks denote the names of new genera
In the subfamily Densovirinae, attributable sequences are available only for a small number of economically significant viruses, which is likely to reflect poorly the diverse nature of viruses infecting hosts from the immense phylum Arthropoda. Accordingly, the >30 % identity requirement is applied less rigorously within this subfamily, in order to allow clustering of monophyletic viruses with conspicuously similar characteristics from host orders separated by large evolutionary distances. Constituent viruses in most of these genera are closely related and infect arthropods from the same host order. However, in the proposed genus Ambidensovirus, certain pairs of viruses that infect different orders of hosts fall short of the proposed identity requirements. Nevertheless, all of these viruses exhibit a complex genomic rearrangement that allows them to co-ordinate bidirectional transcription, which is not seen in viruses from any other parvovirus taxon. In the existing taxonomy, most of these “ambisense” viruses cluster in the genus Densovirus, with a single outlier (Periplaneta fuliginosa densovirus) that infects a blattodean host and is the sole member of genus Pefudensovirus. Recently, this organization was challenged by the identification of four new isolates with ambisense organization, which infect insects from different host orders and are closely related to, albeit not monophyletic with, viruses from both of the existing ambisense genera. To resolve this situation, it is proposed that the six groups should be combined as distinct species in a single monophyletic Ambidensovirus genus, which will have slightly relaxed demarcation criteria that likely reflect the host diaspora. Members of the proposed genus Ambidensovirus thus appear to illustrate how host divergence may mask and complicate underlying sequence-based phylogeny: using current approaches, it would prove challenging to track viral lineages with less conspicuous genomic rearrangements against this background of host-related genetic drift.
Previously, species in the family were generally required to be >95 % related in the NS1 DNA sequence, which is so high a level that many current species consist of single isolates. This has fuelled confusion between taxa and viruses in the literature, and it allows the species level to contribute little to taxonomic structure. The proposals will decrease species identity criteria significantly, requiring viruses in a species to encode NS1 proteins that show >85 % amino acid sequence identity while diverging by >15 % from viruses in other species. This adjustment permits a species to contain a greater diversity of viruses than is currently the case, so that it typically designates a distinct phylogenetic branch and thus adds useful structure within the genus. Other existing criteria, such as host, antigenic properties, and genome characteristics, are still considered.
The two subfamilies, Parvovirinae and Densovirinae, are distinguished primarily by their respective ability to infect vertebrate and arthropod hosts, and this remains the case in the proposals. This separation is supported by Bayesian phylogeny, although it is not immediately apparent under the rooting procedure used in Fig. 1.
Taxon nomenclature
Systematic changes are proposed at the level of species, in part because decreasing the mandatory level of sequence identity for this taxon effectively does away with current species divisions. The call for new names that encompass broader groups of viruses thus provides an opportunity for the field to adopt a non-Latinized, binomial system that has been discussed extensively in the literature [15, 16] and is commonly in use in other viral families. In the proposed nomenclature, species names are emphatically different from virus names and typically consist of a host taxon, a genus affiliation, and a distinguishing numerical or letter suffix, for example, Rodent protoparvovirus 1 (type species of the genus Protoparvovirus, which includes both the existing type species, Minute virus of mice, and a group of closely related rodent viruses, as detailed in Table 2). Since these names indicate the range of viruses included and their branch within the family, they provide useful information about the likely properties of the virus, and allow for facile addition of new species by simply advancing the numerical suffix, as in Rodent protoparvovirus 2, which currently contains a single virus, rat parvovirus 1. In addition, because the proposed species distinguish major branches within each genus, they will provide taxonomic names for groups of viruses that are now commonly discussed together in the literature. The only exceptions to this standard naming pattern involve two species from the genus Dependoparvovirus, which contain viruses that show excellent potential for clinical use as gene therapy delivery vectors. Viruses in one species are named “adeno-associated virus” plus a hyphenated numeral between 1-4 or 6-13, with individual isolates showing important differences in receptor-binding and tissue-specific transduction efficiency. Because these virus names are so well recognized both inside and outside the field, and because their specific numerals have such important implications, the SG considered it unwise to introduce host taxa or additional numerals into the species name. Accordingly, the proposed name for this species is Adeno-associated dependoparvovirus A (instead of the more systematic Primate dependoparvovirus 1). A second species in this genus is named Adeno-associated dependoparvovirus B and includes one virus, adeno-associated virus-5, which is also of current interest for gene therapy applications.
In general, host taxon descriptors at the level of order, rather than family, are selected for species names to accommodate potential host-range disparity among viruses. However, where such host names were considered confusing or awkward to pronounce, less rigid terms were preferred, as in the use of “pinniped” (fin-footed mammals, including walrus, seals, and sea lions) instead of “carnivore” for viruses of the Californian sea lion, and “ungulate” (hoofed animals) instead of “artiodactyl” for viruses of cows, pigs, and sheep.
The proposals also expand the names of most existing genera by introducing an affix into each name. Two distinct problems are addressed in this way. First, it requires specialist knowledge to recognize that Amdovirus, Bocavirus, Dependovirus, and Erythrovirus are genera within one subfamily of the family Parvoviridae, and that Iteravirus is a genus in the subfamily Densovirinae. This dislocation will be addressed by adding the infixes “parvo” or “denso” to indicate subfamily affiliation, as in the genera Amdoparvovirus, Bocaparvovirus, Dependoparvovirus, Erythroparvovirus, and Iteradensovirus. One remaining genus in the subfamily Densovirinae, Brevidensovirus, already contains the infix, and proposed names for all new genera will include the appropriate notation. It is hoped that this modification will improve family recognition, thus providing information about the general properties of a virus in any given genus to people outside the field, and will obviate the need to explain the taxonomy whenever viruses in different parvovirus genera are compared. Practically, it was becoming challenging to invent names for new genera, since these commonly appeared to suggest affiliation to a different virus family. For example, a previously proposed genus name Partetravirus, which is widely in use in the field to encompass viruses related to human parvovirus 4 (PARV4, GenBank AY622943), was not welcomed by the ICTV because it arguably suggested that these viruses were members of the family Alphatetraviridae. In the current proposals, we again seek recognition for this group of viruses, but under the genus name Tetraparvovirus, since the infix should substantially limit ambiguity.
Although subfamily affiliations of viruses in the existing genera Parvovirus and Densovirus are explicit, vernacular use of “parvovirus” and “densovirus” is ambiguous because the terms indicate multiple taxa. Thus, “parvovirus” can refer to members of the genus Parvovirus, the subfamily Parvovirinae, or the family Parvoviridae, while “densovirus” can indicate genus or subfamily affiliation. To provide greater taxonomic precision, the proposals also insert the prefix “Proto” before Parvovirus, creating the genus name Protoparvovirus (from Greek, “proto” meaning “first”, in this case the first viruses identified), and “Ambi” before Densovirus, creating the genus name Ambidensovirus (from Latin or Celtic, meaning “both”, referring to ambisense transcription). Overall, these changes should provide the field with a more self-explanatory framework and greater precision when using taxonomically derived terms.
Taxon and virus lists for the proposed classification are shown in Tables 2 and 3. In the subfamily Parvovirinae, there are three new genera, to be called Aveparvovirus, to indicate the bird (Aves) hosts of the founding members, Copiparvovirus, a siglum for cow and pig, which were the hosts of the first two species identified, and Tetraparvovirus, from the name of the founder virus, human parvovirus 4 (PARV4), using Latin “tetra” in place of the numeral 4. In the subfamily Densovirinae, two new genera are proposed, in order to accommodate shrimp viruses. These will be called Hepandensovirus, to reflect the original name of these viruses, “hepatopancreatic parvovirus”, and Penstyldensovirus, a siglum for Penaeus stylirostris, the host, and name, of the founding member of this species.
As a general rule, the proposals do not tamper with existing viral names, which remain written in Roman script, for example, canine minute virus and Galleria mellonella densovirus (in this case capitalized because “Galleria” is derived from a formal name), whereas all formal taxonomic names, for family, subfamily, genus, and species, are capitalized and written in italics. Although abbreviations of viral names also have no formal standing, those listed in Tables 2 and 3 are recommended by the SG, in order to encourage uniformity. For viruses from the subfamily Densovirinae, viral names have typically been assembled from binomial host names plus the word “densovirus”, for example, Jujonia coenia densovirus, originally abbreviated to JcDNV (where the capitalized “N” harks back to a time when these viruses were called “densonucleosis viruses”). However, many host species share the same initials, and viruses from multiple densovirus genera can infect a single host species. Therefore, as new viruses were identified, their abbreviations were distinguished from pre-existing isolates by the insertion of additional letters, causing them to become progressively longer. In part to offset this continued expansion, the SG suggest eliminating the vestigial N from all abbreviations, as implemented in Table 3. Finally, the proposed establishment of two new genera for shrimp viruses, each encompassing viruses that are responsible for an economically significant disease but which all infect an overlapping group of host species, was deemed to require an unusual approach. As discussed above, one of these viral clusters, formerly known as “hepatopancreatic parvovirus” of shrimp (HPV), now constitutes the genus Hepandensovirus, whereas the other, formerly known as “infectious hypodermal and hematopoietic virus“ of shrimp (IHHNV), is classified in the genus Penstyldensovirus. Because these names and abbreviations do not meet standard densovirus conventions, in this particular instance the SG voted to rename the viruses. However, rather than use “densovirus”, the new genus name was included, in order to improve clarity. Accordingly, in Table 3 these viruses are called, for example, Penaeus monodon hepandensovirus 1-4, or Penaeus monodon penstyldensovirus 1-2, and are abbreviated to PmoHDV (1-4) and PmoPDV (1-2), respectively.
References
Belyi VA, Levine AJ, Skalka AM (2010) Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: the parvoviridae and circoviridae are more than 40 to 50 million years old. J Virol 2010(84):12458–12462
Cotmore SF and Tattersall P. 2013 Parvovirus diversity and DNA damage responses. Cold Spring Harb Perspect Biol. 2013 5(2). doi:10.1101/cshperspect.a012989
Cotmore SF, Tattersall P (2005) A rolling-hairpin strategy: basic mechanisms of DNA replication in the parvoviruses. In: Kerr J, Cotmore SF, Bloom ME, Linden RM, Parrish CR (eds) Parvoviruses. Hodder Arnold, London, pp 171–181
Drummond AJ, Ho SYW, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Eddy SR (2011) Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195
Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, Shi Z (2012) Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol 86:4620–4630
Huang Q, Deng X, Yan Z, Cheng F, Luo Y, Shen W, Lei-Butters DC, Chen AY, Li Y, Tang L, Söderlund-Venermo M, Engelhardt JF, Qiu J (2012) Establishment of a reverse genetics system for studying human bocavirus in human airway epithelia. PLoS Pathog 8:e1002899
Kapoor A, Simmonds P, Lipkin WI (2010) Discovery and characterization of mammalian endogenous parvoviruses. J Virol 84:12628–12635
Li L, Cotmore SF, Tattersall P (2013) Parvoviral left-end hairpin ears are essential during infection for establishing a functional intranuclear transcription template and for efficient progeny genome encapsidation. J Virol 87:10501–10514
Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequences of two proteins. J Mol Biol 48:443–453
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony method. Mol Biol Evol 28:2731–2739
Tattersall P, Bergoin M, Bloom ME, Brown KE, Linden RM, Muzyczka N, Parrish CR, Tijssen P (2005) Family Parvoviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds) Virus taxonomy—eighth report of the International Committee on Taxonomy of viruses. Elsevier/Academic Press, San Diego, pp 353–369
Tijssen P, Agbandje-McKenna M, Almendral JM, Bergoin M, Flegel TW, Hedman K, Kleinschmidt J, Li Y, Pintel DJ, Tattersall P (2011) The family Parvoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy—Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier/ Academic Press, London, pp 405–425
van Regenmortel MHV, Mahy BWJ (2004) Emerging issues in virus taxonomy. Emerg Infect Dis 10:8–13
van Regenmortel MH, Burke DS, Calisher CH, Dietzgen RG, Fauquet CM, Ghabrial SA, Jahrling PB, Johnson KM, Holbrook MR, Horzinek MC, Keil GM, Kuhn JH, Mahy BW, Martelli GP, Pringle C, Rybicki EP, Skern T, Tesh RB, Wahl-Jensen V, Walker PJ, Weaver SC (2010) A proposal to change existing virus species names to non-Latinized binomials. Arch Virol 155:1909–1919
Acknowledgments
This work was supported in part by Public Health Service grants from the National Institutes of Health (CA029303 and AI026109), SFC and PTa; (AI046458 and AI091588), DJP; (AI070723), JQ; (GM082946), MAM; the National Science Foundation (MCB 0718948), MAM; the UK Medical Research Council, AD and DG; the Helsinki University Research Funds and Jusélius Foundation, Finland, MSV; and the Natural Sciences and Engineering Research Council of Canada, PTi.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is related to an ongoing taxonomic proposal, submitted to the International Committee on Taxonomy of Viruses (ICTV) and still under deliberation. The taxonomic changes discussed here may differ from any new taxonomy that is ultimately approved.
S. F. Cotmore, M. Agbandje-McKenna, J. A. Chiorini, D. V. Mukha, D. J. Pintel, J. Qiu, M. Soderlund-Venermo, P. Tattersall and P. Tijssen are the members of the ICTV Parvoviridae Study Group.
Rights and permissions
About this article
Cite this article
Cotmore, S.F., Agbandje-McKenna, M., Chiorini, J.A. et al. The family Parvoviridae . Arch Virol 159, 1239–1247 (2014). https://doi.org/10.1007/s00705-013-1914-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1914-1