
Abstract
Parkinsonism secondary to viral infections is not an uncommon occurrence and has been brought under the spotlight with the spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. A variety of viruses have been described with a potential of inducing or contributing to the occurrence of parkinsonism and Parkinson’s disease (PD), although the relationship between the two remains a matter of debate originating with the description of encephalitis lethargica in the aftermath of the Spanish flu in 1918. While some viral infections have been linked to an increased risk for the development of PD, others seem to have a causal link with the occurrence of parkinsonism. Here, we review the currently available evidence on viral-induced parkinsonism with a focus on potential pathophysiological mechanisms and clinical features. We also review the evidence on viral infections as a risk factor for developing PD and the link between SARS-CoV-2 and parkinsonism, which might have important implications for future research and treatments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Infectious causes for movement disorders are not a rare occurrence and are among the most common causes of secondary movement disorders, with dystonia and parkinsonism being the most prevalent forms (Netravathi et al. 2012). While the former tends to be the dominant type of secondary movement disorder observed in children, the latter becomes more prevalent in adults, suggesting that age at insult influences the clinical picture (Netravathi et al. 2012). The relationship between viruses and parkinsonism has been recently brought under the spotlight due to the ongoing Coronavirus Disease 2019 (COVID-19) pandemic (Ghosh et al. 2021a). The reason behind such interest has historical roots in the observations relating to the ‘encephalitis lethargica’ and post-encephalic parkinsonism, which was linked to the Influenza A virus subtype H1N1 pandemic which occurred over a century ago (Taubenberger 2006; Xing et al. 2022). In addition, exposure to other viruses is known to be associated with an increased risk for the development of Parkinson’s disease (PD); these include influenza, herpes simplex, and hepatitis B (HBV) and C virus (HCV) as shown in a recent meta-analysis (Wang et al. 2020). In this review, we summarise the available evidence on viral-induced parkinsonism with a focus on postulated pathophysiological mechanisms and clinical features. We also review the evidence on viral infections and risk of developing PD.
Viruses and parkinsonism
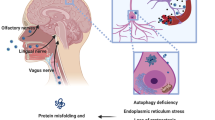
Viral infections can induce parkinsonism by immediate or delayed mechanisms of damage resulting in para-infectious and post-infectious forms of parkinsonism, respectively (Fig. 1 and Table 1). Of note, the two types of damage can often coexist (Sulzer et al. 2020).

Putative pathophysiological mechanisms underlying viral infection-induced parkinsonism
The term “para-infectious” indicates a clinical event that occurs within 15 days from the infectious episode. A para-infectious parkinsonism might be underpinned by direct or indirect pathophysiological mechanisms (Bozzola et al. 2021). Viruses can be neurotropic and directly damage the nigrostriatal pathway accessing the central nervous system (CNS) via three possible routes of entry: 1. peripheral nerves; 2. blood–brain barrier (BBB); 3. blood-cerebrospinal fluid barrier (Fig. 1). Moreover, a viral infection can indirectly damage the nigrostriatal pathway via inducing inflammatory, vascular and/or hypoxic injury (Fig. 1).
Post-infectious parkinsonisms are thought to occur through a pathogen-induced autoimmunity. This may be the result of a non-specific immune activation or due to a targeted immune response to a specific host antigen. Furthermore, persistent infections may continue to drive immune responses leading to chronic inflammation or development of autoimmune processes, resulting in chronic immune stimulations that drive immune-mediated neurologic complications and damage to the nervous system (Johnson and Nath 2018). Of note, viruses can trigger autoimmune reactions through multiple mechanisms including molecular mimicry, bystander activation, and viral persistence with or without epitope spreading (Fig. 1). Molecular mimicry occurs when structural similarities between viral and host antigens trigger activation of T or B cell responses targeting both host and auto-antigens (Blackburn and Wang 2020), for example in the case of Herpes simplex virus 1 (HSV-1) and Epstein-Barr virus (EBV) (Caggiu et al. 2016, 2017; Woulfe et al. 2000). In addition, virus-specific T cells can initiate a bystander activation. Once the virus-specific T cells encounter virus-infected cells, the latter present viral peptides in the context of MHC human leukocyte antigen (HLA) class I or class II molecules to virus-specific CD8 + or CD4 + T cells respectively. This is followed by release of cytokines such as tumour necrosis factor (TNF), lymphotoxin (LT), and nitric oxide (NO), which can lead to bystander killing of the uninfected neighbouring cells (Fujinami et al. 2006). Epitope spreading is a phenomenon related to the bystander activation, which further propagates the inflammatory response. The initial immune response to an antigen is limited to particular peptide sequences on the targeted protein. Subsequently, different epitopes on the inciting antigen, or wholly distinct antigens are involved in the response, which is described as epitope spreading (Blackburn and Wang 2020). Persistent infection, in turn, leads to polyclonal proliferation of B and T cells thereby contributing to autoimmunity (Blackburn and Wang 2020). Virus-specific mechanisms of entry into the CNS and damage are described in the following sections.
Human immunodeficiency virus (HIV)
Human immunodeficiency virus (HIV) is a retrovirus that has been shown to be the cause of acquired immunodeficiency syndrome (AIDS) (Gallo et al. 1984). An estimated 37.9 million individuals are living with HIV worldwide. Infection with HIV results in a failure of the immune system, leading to life-threatening opportunistic infections. The predominant cellular lesion in HIV infection is a progressive loss of CD4+ T cells whose levels correlate with the viral load (Ghosn et al. 2018). CNS involvement can occur quickly as HIV may enter the nervous system within the first weeks after the infection (Schacker et al. 1996; Pilcher et al. 2001).
The mechanisms of HIV entry into the brain include the ‘Trojan horse’ model (migration of the virus across the BBB via infected monocytes or lymphocytes) or the free virions model (through infected endothelial cells) (Ene 2018). Once in the brain, HIV has been clearly shown to infect astrocytes, microglia (Budka 1990; Brack-Werner 1999) and neurons (Wheeler et al. 2006; Cantó-Nogués et al. 2005). It has been proposed that HIV infection of monocytes and macrophages leads to the production of neurotoxins. These putative toxins include HIV proteins like gp120 and tat, cytokines, NO, glutamate, and quinolinic acid (Koutsilieri et al. 2002; Lopez et al. 1999; Sardar et al. 1996). HIV proteins gp120 and tat have been linked with neuronal injury in a series of experiments (Lipton 1992). Subcortical involvement, including involvement of the basal ganglia, has been demonstrated in HIV infection. Autopsy findings by Navia et al. in patients with HIV-associated dementia showed microglial nodule encephalitis with multinucleated giant cells, particularly involving the putamen and caudate nuclei (Navia et al. 1986). Autopsy studies in clinically asymptomatic patients by Reyes et al. showed nigral degeneration with neuronal loss, extracellular melanin and reactive astrocytosis (Reyes et al. 1991).
Patients with AIDS may develop parkinsonism as part of an HIV-related encephalopathy associated with severe damage to the dopaminergic basal ganglia (Clifford 2000). Exposure to antidopaminergic drugs can often trigger an akinetic rigid syndrome due to the state of vulnerability unleashed by the nigral degeneration due to HIV neuroinvasion (Tse et al. 2004). Rarely, parkinsonism can be the presentation of an underlying opportunistic infection (Carrazana et al. 1989). Indeed, parkinsonism has also been reported in association with progressive multifocal leukoencephalopathy (Werring and Chaudhuri 1996) and CNS tuberculosis. HIV-related parkinsonism is typically early-onset (Limphaibool et al. 2019), non–levodopa-responsive, and has often an atypical presentation with symmetrical bradykinesia and rigidity. Rest tremor is usually absent and postural tremor is prominent with early presentation of postural instability and gait difficulties. The parkinsonism observed in patients with AIDS is often associated with dementia, seizures, vacuolar myelopathy and peripheral neuropathy (Mirsattari et al. 1998; Mattos et al. 2002). Primary HIV-associated parkinsonism often appears within several months of HIV infection and is associated with a poor prognosis (Mattos et al. 2002). Diffuse nigral degeneration with loss of both pigmented and non-pigmented neurons, extracellular melanin and reactive astrocytosis have been well-documented even in clinically asymptomatic AIDS patients (Reyes et al. 1991) with a pattern of degeneration that is quite different from the predominantly ventro-lateral nigral neuronal loss seen in idiopathic PD (Itoh et al. 2000). The nigral neuroinvasive potential could explain both the occurrence of parkinsonism and the susceptibility to develop a drug-induced parkinsonism. The management of patients with HIV who present with parkinsonism involves identification and treatment of opportunistic infections, careful review of the patient’s medications for potential extrapyramidal side effects and the use of highly active antiretroviral therapy (HAART). Symptomatic treatment of the movement disorder is often unsatisfying. Evidence suggests that HAART may be helpful in the control and prevention of parkinsonsm in patients who are HIV positive (Rosso et al. 2009).
West Nile virus (WNV)
West Nile virus (WNV) is a mosquito-borne flavivirus and human neuropathogen that has been implicated in several outbreaks in the last two decades (Campbell et al. 2002). The incubation period usually ranges from 2 to 14 days (Zou et al. 2010). Most human infections are subclinical or manifest as a mild febrile illness, but 1% of patients may develop an acute neurologic syndrome, including meningitis, encephalitis, acute flaccid paralysis, and movement disorders (parkinsonism, tremor, opsoclonus-myoclonus, chorea, ataxia, and myoclonus) (Lenka et al. 2019). Neurological complications are more frequently observed in the elderly, alcoholics, and transplant recipients (Lenka et al. 2019).
After the mosquito bite, viral replication occurs in lymph nodes and the spleen, following which viral dissemination occurs in the bloodstream. Although the precise mechanism for WNV entry into the CNS has not been delineated, preclinical studies suggest that Toll-like receptor 3 (TLR-3) and peripheral production of TNF‐α may lead to breakdown of the BBB, thereby facilitating viral entry into the CNS (Wang et al. 2004). Samuel et al. demonstrated retrograde axonal transport through the peripheral neurons, thereby leading to CNS invasion in hamster models (Samuel et al. 2007). Several other mechanisms of CNS entry, including endothelial cell infection and migration into the brain parenchyma via WNV-infected leucocytes migrating into CNS (Sejvar 2016), have been postulated. Neuronal infection is followed by cell death. Histopathologic findings in humans are characterized by the presence of microglial nodules composed of lymphocytes and histiocytes, while leptomeningeal mononuclear inflammatory infiltrates are present in cases of meningitis. CD8 T-lymphocytes represent the predominant inflammatory cell type in the nodules and infiltrates (Sampson et al. 2000; Doron et al. 2003).
The emergence of parkinsonism in WNV infection is likely secondary to the specific neurotropism of WNV for deep gray matter nuclei, especially the substantia nigra (Lenka et al. 2019; Bosanko et al. 2003; Solomon et al. 2003). Parkinsonian features are usually self-limiting, but tremor persists for a long time after the infection in 10% of the cases with subsequent disability (Patel et al. 2015). Brain MRI may show abnormalities in the basal ganglia, thalamus, and pons, mostly bilaterally, evident in T2 and DWI sequences (Sejvar 2014). WNV infection is most sensitively assessed by the presence of immunoglobulin M (IgM) antibody in the CSF (Solomon et al. 2003). To date, no specific treatment for WNV other than symptomatic measures are available.
Japanese encephalitis B virus (JEV)
Japanese encephalitis virus (JEV) is a flavivirus causing a mosquite-borne disease which is endemic in Southeast Asia and representing the most common cause of encephalitis (Diagana et al. 2007). Unlike other flaviviruses, JEV leads to frequent and severe neurological complications (movement disorders, poliomyelitis-like flaccid paralysis, seizures, Guillain-Barré syndrome, etc.), often leading to severe disability, while systemic features including haemorrhage and jaundice are infrequent (Turtle and Solomon 2018). JEV affects mostly children in rural areas. (Turtle and Solomon 2018)
JEV enters the bloodstream through the bite of Culex mosquito (Diagana et al. 2007). Subsequently, binding to the CNS vascular endothelial cells allows entry into CNS; BBB disruption ensues owing to the host inflammatory response (Hsieh et al. 2019). Kalia et al. demonstrated that the mechanism of entry of JE virions into neuronal cells involves dynamin and plasma membrane cholesterol (Kalia et al. 2013). Direct neuronal damage owing to viral replication within neuronal cells and an inflammatory response have been linked to neuronal apoptosis (Raung et al. 2001). Interleukin 6 (IL-6), TNF‐α, monocyte chemotactic protein 1 (MCP1), and Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted (RANTES) are thought to mediate the inflammatory response (Ghoshal et al. 2007; Chen et al. 2004).
JEV targets dopaminergic neuron-rich areas of the brain such as thalamus and midbrain. JEV-infected Fisher rats showed profound gliosis in the substantia nigra pars compacta (SNpc), similar to that seen in PD (Ogata et al. 1997). Hamue et al. reported reduced norepinephrine and dopamine levels in rats infected by JEV (Hamaue et al. 2006). Misra et al. described lower CSF concentrations of dopamine, norepinephrine and homovanillic acid, in patients with JE-associated movement disorders, stemming from impairment of the dopaminergic and norepinephrinergic systems (Misra et al. 2005).
JEV infection begins as an undifferentiated febrile illness (Turtle and Solomon 2018). If the encephalitic syndrome occurs, the brain regions noted to become involved include the thalamus, basal ganglia, brain stem, cerebellum, hippocampus, and cerebral cortex (Ishii et al. 1977). Parkinsonism is observed in a significant proportion of patients 2–6 weeks after the acute encephalitic phase (Misra and Kalita 1997), characterized by bradykinesia, rigidity, prominent masked facies and hypophonia. JEV-related parkinsonism tends to regress in most in 1–3 weeks although some patients manifest permanent sequelae, especially hypophonia (Murgod et al. 2001; Misra and Kalita 2002). Neuroimaging in patients with JEV infection may show lesions in the substantia nigra and typical extensive bilateral thalamic involvement, with a propensity for petechial haemorrhages (Murgod et al. 2001; Kumar et al. 1997). To date, no specific treatment for Japanese encephalitis other than symptomatic measures has been shown to work (Turtle and Solomon 2018).
Influenza and the case of Encephalitis Lethargica/post-encephalitic Parkinsonism
The causative association between parkinsonism and infections may be very intricate. While many viruses could cause a para-infectious parkinsonian syndrome for their predilection for basal ganglia and substantia nigra neurotropism, as explained in the previous sections, the pathogenic mechanisms of a post-infectious parkinsonism, whose paradigm is post-encephalitic parkinsonism (PEP), remain elusive. PEP has been historically linked to encephalitis lethargica (EL) and, together, they have been defined as one of the biggest medical mysteries (Hoffman and Vilensky 2017). EL was a polymorphic epidemic neurological syndrome which spread across Europe and then the world in the early part of the twentieth century. Firstly described by von Economo in 1917 (Hoffman and Vilensky 2017; Vilensky et al. 2010a), EL was clinically characterized by flu-like symptoms, sleepiness, disorders of ocular motility, fever, and movement disorders (both hypo- and hyperkinetic), although virtually any neurological sign or symptom could be exhibited. PEP is considered to have occurred months to years later and was most commonly characterized by akinetic-rigid features. Interestingly, decades later some of these individuals responded clinically to levodopa (Hoffman and Vilensky 2017). In the decades following the epidemic, it was estimated that as many as 50% of parkinsonian cases were postencephalitic (Krusz et al. 1987). Neuropathology of postencephalitic parkinsonism shows diffuse brain atrophy and marked neuronal loss and gliosis particularly in the substantia nigra and, interestingly, a distribution of neurofibrillary tangles that is nearly identical histopathologically to what can be observed in other neurodegenerative parkinsonisms (Jellinger 2009). The aetiology of this clinical entity is unknown, but it is generally assumed that, considering the influenza-like prodrome of the acute phase and the contemporaneous Spanish H1N1 influenza A pandemic, EL had a viral aetiology (Vilensky et al. 2010a). In fact, the 1918 flu pandemic affected large parts of the world population and is thought to have killed at least 40 million people during the same decade (Reid et al. 2001). Although Gamboa et al. have demonstrated the presence of viral antigens in the hypothalamus and midbrain of patients with PEP (Gamboa et al. 1974), other authors failed to identify the presence of the virus (Berger and Vilensky 2014). Interestingly, it has been demonstrated that several types of influenza A virus can travel into the nervous system following a systemic infection (Tanaka et al. 2003), can localize selectively in the ventral substantia nigra and hippocampus (Takahashi et al. 1995) and can induce neuroinflammation, protein aggregation and degeneration of dopaminergic neurons in the substantia nigra pars compact (SNpc) (Jang and Dezzutti 2009). Although the dopaminergic neuronal perturbation appears to be transient, the inflammatory response with permanent activation of microglia seems to persistas a long-term effect (Jang et al. 2012). Recent studies have questioned the simplistic assertion that EL led directly to PEP (Vilensky et al. 2010a, b) and have reconsidered the causative role of an infection in the context of an autoimmune disorder that could have been triggered by an infectious agent (Dale et al. 2004).
Other post-viral parkinsonism
A number of viruses have been associated with both acute and chronic parkinsonism. Apart from the above-mentioned viral agents, other viruses associated with parkinsonism are Coxsackie (Walters 1960; Poser et al. 1969), Western equine encephalitis (Mulder et al. 1951), EBV (Espay and Henderson 2011; Dimova et al. 2006), Cytomegalovirus (Giraldi et al. 1991), St. Louis virus (Pranzatelli et al. 1994), Herpes simplex virus (Ickenstein et al. 1999), and Poliovirus (Vincent and Myers 1978).
Viruses and idiopathic Parkinson’s disease
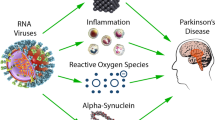
Several viral infections have been associated to an increased risk of developing PD (Smeyne et al. 2020) and the observation of PD-like symptoms in individuals following viral infections (see paragraph Viruses and parkinsonism above) has led to the hypothesis of a contributory role of viral infections in the pathogenesis of idiopathic PD. Evidence supporting this hypothesis comes from preclinical studies looking at the potential associations between α-synuclein accumulation and viral infections, as well as from epidemiological studies reporting increased risk of developing idiopathic PD following a viral infection.
Among these, influenza viruses are among those with the most robust evidence. In a study conducted on human mesencephalic dopaminergic cells in vitro and in Rag knockout mice in vivo, it has been shown that acute H1N1 infection led to the formation of α-synuclein aggregates through a H1N1-induced blocking of autophagosome formation and inhibition of autophagic flux (Marreiros et al. 2020). This would suggest that an aberrant proteostasis induced by the virus may play a role in the initiation of the protein misfolding process. Of note, it has been shown that treatment with oseltamivir phosphate, an anti-influenza compound, prevented H1N1-induced α-synuclein aggregation, and vaccination or treatment with oseltamivir carboxylate improved the dopaminergic neuron integrity in a model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated animals infected with H1N1 (Marreiros et al. 2020; Sadasivan et al. 2017). An earlier study conducted on a mouse model infected by the H5N1 avian influenza virus showed that the virus could travel from the peripheral into the CNS, causing activation of microglia together with α-synuclein phosphorylation and aggregation in the infected regions, which persisted long after the resolution of the infection (Jang et al. 2009). In addition, a significant loss of dopaminergic neurons in the SNpc was found 60 days after the infection (Jang et al. 2009). Epidemiological studies have provided support to the possible pathogenic role of influenza viruses in idiopathic PD, although with some controversies. While a UK community-based study conducted in 2009 found an association between influenza diagnosis and PD-like symptoms but not development of idiopathic PD (Toovey et al. 2011), a subsequent population-based Canadian study found that severe influenza was associated with subsequent development of idiopathic PD (odds ratio: 2.01; 95% confidence interval: 1.16–3.48), although the association was less significant for infections occurred more than 10 years before PD diagnosis (Harris et al. 2012). Interestingly, in a recent Danish case–control study influenza was significantly associated with diagnoses of PD more than 10 years after infection, while other infections did not show such association, suggesting that a long-term association, although not proving causality, may strengthen the hypothesis of the role of influenza virus in contributing or triggering PD-related neurodegeneration (Cocoros et al. 2021).
Hepatitis virus infection has also been proposed as a potential pathophysiological factor in PD. HCV, in particular, has been linked with idiopathic PD in several epidemiological studies (Wu et al. 2015; Tsai et al. 2016; Pakpoor et al. 2017; Goldstein et al. 2019; Choi et al. 2020). Two meta-analyses (Wang et al. 2020; Wijarnpreecha et al. 2018) have demonstrated an increased risk of PD among HCV-infected patients. Interestingly, Su et al. (2019) found that PegIFN/RBV antiviral therapy was associated with a reduced risk of parkinsonism or PD in a cohort of patients with chronic HCV. Of note, Goldstein et al. (2019) found an increased risk for PD also in the group of patients with non-alcoholic steatohepatitis, arguing that liver disease per se could be a risk factor for PD rather than the viral infection and that a possible misclassification of cirrhosis-induced parkinsonism as PD could at least partially explain the association between HCV and PD. However, the mechanisms behind the link between HCV and PD is still unclear. In a study conducted by Wu et al. (2015) on midbrain neuron-glia coculture system in rats, HCV infection induced 60% of dopaminergic neuron death, similar to that of MPTP, suggesting the massive release of inflammatory cytokines might be the trigger for the observed PD-related pathological changes. While the neurotropism of HCV has now been established (Fletcher et al. 2012), it is not clear, however, if the neuronal damage could be caused by the viral replication at the CNS level or by the immune response of the infected cells (Amor et al. 2010). In addition, alteration of striatal dopaminergic transporter binding has been observed in HCV-infected patients with chronic fatigue and cognitive impairment, suggesting a defective dopaminergic transmission that could potentially underlie the association between HCV and PD (Weissenborn et al. 2006). Whether there is an association between HBV and PD is still uncertain, with some epidemiological studies reporting an increased risk of PD in HBV-infected individuals (Pakpoor et al. 2017; Choi et al. 2020) and others reporting inconsistent results (Wu et al. 2015; Tsai et al. 2016; Goldstein et al. 2019; Wang et al. 2020).
Studies have shown that EBV, a double-stranded DNA virus, can replicate in the CNS and disrupt the integrity of the BBB, causing neuronal damage and inflammation (Gent et al. 2014; Liu and Cohen 2016). It has been demonstrated that patients with PD are significantly more seropositive for EBV than the general population (Bu et al. 2015). Furthermore, it has been hypothesized that latent EBV infection can trigger autoantibodies targeting the critical repeat region cross-react with the homologous epitope on α-synuclein and induce its oligomerization (Woulfe et al. 2000).
While parkinsonism has been recognised as a potential manifestation of HIV-associated encephalopathy, it is still unclear whether HIV infection plays a role in the pathogenesis of idiopathic PD. A recent study by Santerre et al. (2021) investigated the effect of exposure to HIV-1 Vpr protein on human neuroblastoma cells (which share many features with dopaminergic neurons) and it was observed that HIV-1 Vpr protein triggers the accumulation of α-synuclein in neurons after decreasing lysosomal acidification, deregulating lysosome positioning, and the expression levels of several proteins involved in lysosomal maturation, suggesting that the suppression of neuronal autophagy exerted by the viral protein could be a mechanism responsible for toxic protein aggregation and subsequent neurodegeneration.
Caggiu et al. (2016) proposed a mechanism of molecular mimicry between HSV-1 and α-synuclein in membranes of dopaminergic neurons of the SNpc. The antibody response against homologous HSV peptides has been shown to be more prevalent among patients with PD thanin healthy controls. Cross-reactivity has been demonstrated between HSV-1 and human α-synuclein peptides indicating that HSV-1 may play a role in triggering an autoimmune response against the neurones of SNpc in PD. The same peptides are able to induce cell-mediated responses in PD patients highlighting the relevant role of TNF-α and neuroinflammation (Caggiu et al. 2017). Similarly, evidence for molecular mimicry between α-synuclein and viral protein of EBV has been demonstrated, highlighting this autoimmune mechanism in the pathophysiology of PD (Woulfe et al. 2000). A recent Taiwanese population-based cohort study revealed an increased risk of PD development among elderly patients with a prior diagnosis of herpes zoster when compared to those without history of herpes zoster infection (Lai et al. 2017).
COVID-19, parkinsonism and Parkinson’s disease: present and future
PD is a chronic and progressive neurodegenerative disease warranting special consideration during the COVID-19 pandemic due to an increased vulnerability attributed to people with this condition (Lau et al. 2021). In part, this may be due to elderly and advanced-stage people with PD (PwP) developing progressive rigidity of respiratory muscles and the thoracic cavity, in addition to a weakened abnormal posture, as well as an impaired cough reflex (Wamelen et al. 2020; Emmi et al. 2022). In relation to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in PwP, pneumonia was by far the most common cause of inpatient admission, and the main cause of death (Okunoye et al. 2020), especially among older PwP with longer disease duration and those on device-aided therapies showing mortality rates of up to 40% (Antonini et al. 2020). Nonetheless, there seems to be a lack of consensus on the COVID-19 related mortality of PwP (Hippisley-Cox et al. 2021).
While acute implications of SARS-CoV-2 infection in PwP, particularly worsening of motor and non-motor symptoms, have been described in several studies (Antonini et al. 2020; Fearon and Fasano 2021), little is known about possible long term sequelae of COVID-19 in PwP. After the initial wave of COVID-19 pandemic, long-COVID or the “post–COVID-19 syndrome” emerged and is now defined by the National Institute for Health and Care Excellence in the United Kingdom as signs and symptoms that occur during or after an infection consistent with COVID-19, persisting for more than 12 weeks which are not explained by an alternative diagnosis (Sivan and Taylor 2020). According to one multi-centre cohort study, the most common long-term effects of COVID-19 in PwP are worsening of motor function, followed by increased levodopa daily dose requirements, fatigue and cognitive disturbances which includes “brain fog”. Other complications include reduced concentration and memory decline as well as sleep disturbances. However, the authors also suggested that post-COVID clinical features may be due to an effect of quarantine and viral infection causing deterioration of the pre-existing PD features (Leta et al. 2021).
The idea of the COVID-19 pandemic unmasking parkinsonism cases in the near or distant future due to the widespread of the virus globally has recently become a source of speculations and concern to many researchers (Beauchamp et al. 2020; Brundin et al. 2020). Up to now, 20 cases of de novo parkinsonism, presenting during or shortly after a SARS-CoV-2 infection, have been described in the literature (Faber et al. 2020; Méndez-Guerrero et al. 2020; Cohen et al. 2020; Pilotto et al. 2021; Akilli and Yosunkaya 2021; Makhoul and Jankovic 2021; Roy et al. 2021; Fearon et al. 2021; Tiraboschi et al. 2021; Rass et al. 2021; Ghosh et al. 2021b; Ayele et al. 2021; Morassi et al. 2021; Cavallieri et al. 2021; Ong et al. 2022; Rao et al. 2022). None of these patients had a family history of PD (see Table 2).
The majority of these patients would well-fit the concept of post-infectious parkinsonism with relevant symptoms developing either in the context of encephalopathy (11 patients) or without (four patients). Immunomodulatory/immunosuppressive therapy was administered in five of the above cases with four of them exhibiting a good response and authors suggesting an underlying immune-mediated substrate. Interestingly, a case of acute necrotizing encephalopathy (ANEC) has been recognised among these latter cases. Trials of dopaminergic therapy have also been attempted in 11 of the above cases with parkinsonism in most of them responding either significantly (seven patients) or moderately (one patient), thus, suggesting an underlying, occasionally reversible, impairment of the dopaminergic pathway. More specific causes of secondary parkinsonism, precipitated by conditions often accompanying COVID-19, like respiratory impairment or a hypercoagulable state, have also been identified in some of the above occasions, including brain injury due to silent hypoxia (two patients), an acute stroke in the basal ganglia (one patient), and an extra-pontine osmotic demyelination syndrome, triggered by uncontrolled hyperglycemia (one patient). It is also of notice that four patients had a history of neuroleptic drug use prior to the emergence of parkinsonism (either chronic or in the context of COVID-19 hospitalisation to address agitation), thus, the possibility of drug-induced parkinsonism cannot be overlooked.
Considering the remaining patients, the treating physicians have suggested a diagnosis of probable PD in four of them, without, however, being able to exclude a diagnosis of post-infectious parkinsonism. One of these patients mentioned prior symptoms of prodromal PD (constipation), while two patients out of the three patients who were tested, were found positive for a heterozygous mutation in the genes of glucocerebrosidase (GBA) and leucine-rich repeat kinase 2 (LRRK2).
The temporal proximity of a new-onset parkinsonism with a COVID-19 diagnosis, along with concurrent encephalopathy in some cases, have led researchers to suspect an etiological connection between the two. Different mechanisms have been assumed to mediate these cases of supposedly para- or post-infectious parkinsonism, including structural or functional impairment of the nigrostriatal pathway, inflammatory or vascular damage, and unmasking of already active, although asymptomatic, cases of prodromal PD (Merello et al. 2021). However, with more than 5300 confirmed COVID-19 cases per 100,000 globally (as of February 2022) (Worldmeter.info 2021) and an annual incidence of about 15 PD cases per 100,000 (Tysnes and Storstein 2017), anticipating a parkinsonism wave based solely on the current 20 published cases sounds rather premature and susceptible to bias. Although a clear association between COVID-19 and a potential rise in parkinsonism cases cannot be currently justified, greater vigilance is recommended in order to timely recognise and address potential neurological manifestations of COVID-19, including parkinsonism, as the pandemic has only been 2 years in progression and long-term effects might not be evident yet.
Conclusions
Parkinsonism secondary to viral infections is not uncommon and can be mediated by a variety of pathophysiological mechanisms leading to immediate or delayed damage of the nigrostriatal pathways. The available evidence seems to point towards a role of viral infections in the pathogenesis of PD, although this still remains a matter of debate. A clear association between SARS-Cov-2 infection and a rise in parkinsonism cases cannot be currently justified; however, greater vigilance for possible long-term neurological sequalae is encouraged and long-term follow up will provide further information on the association of SARS-CoV-2 with parkinsonism and PD.
References
Akilli NB, Yosunkaya A (2021) Part of the Covid19 puzzle: acute Parkinsonism. Am J Emerg Med 47:333.e1-333.e3. https://doi.org/10.1016/j.ajem.2021.02.050
Boura I, Chaudhuri KR (2022) Coronavirus disease 2019 and related parkinsonism: the clinical evidence thus far. Mov Disord Clin Pract. https://doi.org/10.1002/mdc3.13461
Amor S, Puentes F, Baker D, van der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology 129(2):154–169. https://doi.org/10.1111/j.1365-2567.2009.03225.x
Antonini A, Leta V, Teo J, Chaudhuri KR (2020) Outcome of Parkinson’s disease patients affected by COVID-19. Mov Disord 35(6):905–908. https://doi.org/10.1002/mds.28104
Ayele BA, Demissie H, Awraris M et al (2021) SARS-COV-2 induced Parkinsonism: The first case from the sub-Saharan Africa. Clin Park Relat Disord 5:100116–100116. https://doi.org/10.1016/j.prdoa.2021.100116
Beauchamp LC, Finkelstein DI, Bush AI, Evans AH, Barnham KJ (2020) Parkinsonism as a third wave of the COVID-19 pandemic? J Park Dis 10(4):1343–1353. https://doi.org/10.3233/JPD-202211
Berger JR, Vilensky JA (2014) Encephalitis lethargica (von Economo’s encephalitis). 1ed, vol 123. Elsevier B.V., pp 745–761
Blackburn KM, Wang C (2020) Post-infectious neurological disorders. Ther Adv Neurol Disord 13:1756286420952901
Bosanko CM, Gilroy J, Wang A-M et al (2003) West Nile virus encephalitis involving the substantia Nigra. Arch Neurol 60(10):1448–1448. https://doi.org/10.1001/archneur.60.10.1448
Bozzola E, Spina G, Valeriani M et al (2021) Management of pediatric post-infectious neurological syndromes. Ital J Pediatrics 47(1):17. https://doi.org/10.1186/s13052-021-00968-y
Brack-Werner R (1999) Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS 13(1):1–22. https://doi.org/10.1097/00002030-199901140-00003
Brundin P, Nath A, Beckham JD (2020) Is COVID-19 a perfect storm for Parkinson’s disease? Trends Neurosci 43(12):931–933. https://doi.org/10.1016/j.tins.2020.10.009
Bu XL, Wang X, Xiang Y et al (2015) The association between infectious burden and Parkinson’s disease: a case-control study. Parkinsonism Relat Disord 21(8):877–881. https://doi.org/10.1016/j.parkreldis.2015.05.015
Budka H (1990) Human immunodeficiency virus (HIV) envelope and core proteins in CNS tissues of patients with the acquired immune deficiency syndrome (AIDS). Acta Neuropathol 79(6):611–619. https://doi.org/10.1007/BF00294238
Caggiu E, Paulus K, Arru G, Piredda R, Sechi GP, Sechi LA (2016) Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J Neuroimmunol 291:110–114. https://doi.org/10.1016/j.jneuroim.2016.01.007
Caggiu E, Paulus K, Galleri G et al (2017) Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J Neuroimmunol 310:26–31. https://doi.org/10.1016/j.jneuroim.2017.06.004
Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ (2002) West Nile virus. Lancet Infect Dis 2(9):519–529. https://doi.org/10.1016/s1473-3099(02)00368-7
Cantó-Nogués C, Sánchez-Ramón S, Álvarez S, Lacruz C, Muñóz-Fernández MÁ (2005) HIV-1 infection of neurons might account for progressive HIV-1-associated encephalopathy in children. J Mol Neurosci 27(1):79–89. https://doi.org/10.1385/JMN:27:1:079
Carrazana EJ, Rossitch E Jr, Samuels MA (1989) Parkinsonian symptoms in a patient with AIDS and cerebral toxoplasmosis. J Neurol Neurosurg Psychiatry 52(12):1445–1447. https://doi.org/10.1136/jnnp.52.12.1445-a
Cavallieri F, Fioravanti V, Toschi G et al (2021) COVID-19 and Parkinson’s disease: a casual association or a possible second hit in neurodegeneration? J Neurol. https://doi.org/10.1007/s00415-021-10694-4
Chen CJ, Chen JH, Chen SY, Liao SL, Raung SL (2004) Upregulation of RANTES gene expression in neuroglia by Japanese encephalitis virus infection. J Virol 78(22):12107–12119. https://doi.org/10.1128/jvi.78.22.12107-12119.2004
Choi HY, Mai TH, Kim KA, Cho H, Ki M (2020) Association between viral hepatitis infection and Parkinson’s disease: a population-based prospective study. J Viral Hepatitis 27(11):1171–1178. https://doi.org/10.1111/jvh.13346 (PMID-32558154)
Cocoros NM, Svensson E, Szépligeti SK et al (2021) Long-term risk of Parkinson disease following influenza and other infections. Jama Neurol 78(12):1461–1470. https://doi.org/10.1001/jamaneurol.2021.3895 (PMID-34694344)
Cohen ME, Eichel R, Steiner-Birmanns B et al (2020) A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol 19(10):804–805. https://doi.org/10.1016/s1474-4422(20)30305-7
Dale RC, Church AJ, Surtees RA et al (2004) Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain 127(Pt 1):21–33. https://doi.org/10.1093/brain/awh008
De Mattos JP, De Rosso ALZ, Corrêa RB, Novis SAP (2002) Movement disorders in 28 HIV-infected patients. Arq Neuropsiquiatr 60(3A):525–530. https://doi.org/10.1590/s0004-282x2002000400002
Diagana M, Preux PM, Dumas M (2007) Japanese encephalitis revisited. J Neurol Sci 262(1–2):165–170. https://doi.org/10.1016/j.jns.2007.06.041
Dimova PS, Bojinova V, Georgiev D, Milanov I (2006) Acute reversible parkinsonism in Epstein-Barr virus-related encephalitis lethargica-like illness. Mov Disord 21(4):564–566. https://doi.org/10.1002/mds.20742
Doron SI, Dashe JF, Adelman LS, Brown WF, Werner BG, Hadley S (2003) Histopathologically proven poliomyelitis with quadriplegia and loss of brainstem function due to West Nile virus infection. Clin Infect Dis 37(5):e74–e77. https://doi.org/10.1086/377177
Emmi A, Boura I, Raeder V, Mathew D, Sulzer D, Goldman JE, Leta V (2022) Covid-19, nervous system pathology, and Parkinson's disease: Bench to bedside. Int Rev Neurobiol. https://doi.org/10.1016/bs.irn.2022.06.006
Ene L (2018) Human immunodeficiency virus in the brain-culprit or facilitator? Infect Dis 11:1178633717752687. https://doi.org/10.1177/1178633717752687
Espay AJ, Henderson KK (2011) Postencephalitic parkinsonism and basal ganglia necrosis due to Epstein-Barr virus infection. Neurology 76(17):1529. https://doi.org/10.1212/WNL.0b013e318217e7dd
Faber I, Brandão PRP, Menegatti F, de Carvalho Bispo DD, Maluf FB, Cardoso F (2020) Coronavirus disease 2019 and Parkinsonism: a non-post-encephalitic case. Mov Disord 35(10):1721–1722. https://doi.org/10.1002/mds.28277
Fearon C, Fasano A (2021) Parkinson’s disease and the COVID-19 pandemic. J Parkinsons Dis 11(2):431–444. https://doi.org/10.3233/jpd-202320
Fearon C, Mikulis DJ, Lang AE (2021) Parkinsonism as a sequela of SARS-CoV-2 infection: pure hypoxic injury or additional COVID-19-related response? Mov Disord 36(7):1483–1484. https://doi.org/10.1002/mds.28656
Fletcher NF, Wilson GK, Murray J et al (2012) Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology 142(3):634-643.e6. https://doi.org/10.1053/j.gastro.2011.11.028
Fujinami RS, von Herrath MG, Christen U, Whitton JL (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev 19(1):80–94
Gallo R, Salahuddin S, Popovic M et al (1984) Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224(4648):500–503. https://doi.org/10.1126/science.6200936
Gamboa ET, Wolf A, Yahr MD et al (1974) Influenza virus antigen in postencephalitic parkinsonism brain. Detection by immunofluorescence. Arch Neurol 31(4):228–232. https://doi.org/10.1001/archneur.1974.00490400042003
Ghosh R, Biswas U, Roy D et al (2021a) De Novo movement disorders and COVID-19: exploring the Interface. Mov Disord Clin Pract 8(5):669–680. https://doi.org/10.1002/mdc3.13224
Ghosh R, Ray A, Roy D, Das S, Dubey S, Benito-León J (2021b) Parkinsonism with akinetic mutism following osmotic demyelination syndrome in a SARS-CoV-2 infected elderly diabetic woman: a case report. Neurologia. https://doi.org/10.1016/j.nrl.2021.09.007
Ghoshal A, Das S, Ghosh S et al (2007) Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55(5):483–496. https://doi.org/10.1002/glia.20474
Ghosn J, Taiwo B, Seedat S, Autran B, Katlama C (2018) Seminar HIV. The Lancet 392(18):685–697. https://doi.org/10.1016/S0140-6736(18)31311-4
Giraldi C, Mazzoni M, Morgantini PG, Lunardi GV (1991) Parkinsonian syndrome during viral meningoencephalitis. Riv Neurol 61(5):183–185 (Sindrome parkinsoniana nel decorso di una meningoencefalite virale.)
Goldstein L, Fogel-Grinvald H, Steiner I (2019) Hepatitis B and C virus infection as a risk factor for Parkinson’s disease in Israel—a nationwide cohort study. J Neurol Sci 398:138–141. https://doi.org/10.1016/j.jns.2019.01.012
Hamaue N, Ogata A, Terado M et al (2006) Brain catecholamine alterations and pathological features with aging in Parkinson disease model rat induced by Japanese encephalitis virus. Neurochem Res 31(12):1451–1455. https://doi.org/10.1007/s11064-006-9197-5
Harris MA, Tsui JK, Marion SA, Shen H, Teschke K (2012) Association of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov Disord 27(9):1111–1117. https://doi.org/10.1002/mds.25077 (PMID-22753266)
Hippisley-Cox J, Coupland CA, Mehta N, Keogh RH, Diaz-Ordaz K, Khunti K et al (2021) Risk prediction of covid-19 related death and hospital admission in adults after covid-19 vaccination: national prospective cohort study. BMJ 374:n2244. https://doi.org/10.1136/bmj.n2244
Hoffman LA, Vilensky JA (2017) Encephalitis lethargica: 100 years after the epidemic. Brain 140(8):2246–2251. https://doi.org/10.1093/brain/awx177
Hsieh JT, Rathore APS, Soundarajan G, St John AL (2019) Japanese encephalitis virus neuropenetrance is driven by mast cell chymase. Nat Commun 10(1):706. https://doi.org/10.1038/s41467-019-08641-z
Ickenstein GW, Klotz JM, Langohr HD (1999) Virus encephalitis with symptomatic Parkinson syndrome, diabetes insipidus and panhypopituitarism. Fortschr Neurol Psychiatr 67(10):476–481. https://doi.org/10.1055/s-2007-994998 (Virusenzephalitis mit symptomatischem Parkinsonsyndrome Diabetes insipidus und Panhypopituitarismus)
Clifford DB (2000) Human immunodeficiency virus-associated dementia. Arch Neurol 57(3):321–324. https://doi.org/10.1001/archneur.57.3.321
Ishii T, Matsushita M, Hamada S (1977) Characteristic residual neuropathological features of Japanese B encephalitis. Acta Neuropathol 38(3):181–186. https://doi.org/10.1007/bf00688063
Itoh K, Mehraein P, Weis S (2000) Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol 99:376–384. https://doi.org/10.1007/s004010051139
Jang Y, Dezzutti CS (2009) Viral Parkinsonism look at refernece. Biochim Biophys Acta 18(3):145–150. https://doi.org/10.1038/nbt.3121.ChIP-nexus
Jang H, Boltz D, Sturm-Ramirez K et al (2009) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc National Acad Sci 106(33):14063–14068. https://doi.org/10.1073/pnas.0900096106 (PMID-19667183)
Jang H, Boltz D, McClaren J et al (2012) Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci 32(5):1545–1559. https://doi.org/10.1523/jneurosci.5123-11.2012
Jellinger KA (2009) Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol 118(3):371–379. https://doi.org/10.1007/s00401-009-0537-9
Johnson TP, Nath A (2018) Neurological syndromes driven by postinfectious processes or unrecognized persistent infections. Curr Opin Neurol 31(3):318–324. https://doi.org/10.1097/wco.0000000000000553
Kalia M, Khasa R, Sharma M, Nain M, Vrati S (2013) Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 87(1):148–162. https://doi.org/10.1128/jvi.01399-12
Kobylecki C, Jones T, Lim CK, Miller C, Thomson AM (2020) Phenomenology and outcomes of in-patients with Parkinson's Disease during the coronavirus disease 2019 pandemic. Mov Disord 35:1295–1296. https://doi.org/10.1002/mds.28205
Koutsilieri E, Sopper S, Scheller C, ter Meulen V, Riederer P (2002) Parkinsonism in HIV dementia. J Neural Transm (vienna, Austria : 1996) 109(5–6):767–775. https://doi.org/10.1007/s007020200063
Krusz JC, Koller WC, Ziegler DK (1987) Historical review: abnormal movements associated with epidemic encephalitis lethargica. Mov Disord 2(3):137–141. https://doi.org/10.1002/mds.870020301
Kumar S, Misra UK, Kalita J, Salwani V, Gupta RK, Gujral R (1997) MRI in Japanese encephalitis. Neuroradiology 39(3):180–184. https://doi.org/10.1007/s002340050388
Lai SW, Lin CH, Lin HF, Lin CL, Lin CC, Liao KF (2017) Herpes zoster correlates with increased risk of Parkinson’s disease in older people: a population-based cohort study in Taiwan. Medicine (baltimore) 96(7):e6075. https://doi.org/10.1097/md.0000000000006075
Lau YH, Lau KM, Ibrahim NM (2021) Management of Parkinson’s disease in the COVID-19 pandemic and future perspectives in the era of vaccination. J Mov Disord 14(3):177
Lenka A, Kamat A, Mittal SO (2019) Spectrum of movement disorders in patients with neuroinvasive West Nile Virus infection. Mov Disord Clin Pract 6(6):426–433. https://doi.org/10.1002/mdc3.12806
Leta V, Rodríguez-Violante M, Abundes A et al (2021) Parkinson’s disease and post–COVID-19 syndrome: the Parkinson’s Long-COVID spectrum. Mov Disord 36(6):1287
Limphaibool N, Iwanowski P, Holstad MJV, Kobylarek D, Kozubski W (2019) Infectious etiologies of Parkinsonism: Pathomechanisms and clinical implications. Front Neurol 10(Jun):1–11. https://doi.org/10.3389/fneur.2019.00652
Lipton SA (1992) Models of neuronal injury in AIDS: another role for the NMDA receptor? Trends Neurosci 15(3):75–79. https://doi.org/10.1016/0166-2236(92)90013-x
Liu X, Cohen JI (2016) Epstein-Barr virus (EBV) tegument protein BGLF2 promotes EBV reactivation through activation of the p38 mitogen-activated protein kinase. J Virol 90(2):1129–1138. https://doi.org/10.1128/jvi.01410-15
Lopez OL, Smith G, Meltzer CC, Becker JT (1999) Dopamine systems in human immunodeficiency virus-associated dementia. Neuropsychiatry Neuropsychol Behav Neurol 12(3):184–192
Makhoul K, Jankovic J (2021) Parkinson’s disease after COVID-19. J Neurol Sci 422:117331. https://doi.org/10.1016/j.jns.2021.117331
Marreiros R, Müller-Schiffmann A, Trossbach SV et al (2020) Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci 117(12):6741–6751. https://doi.org/10.1073/pnas.1906466117 (PMID-32152117)
Méndez-Guerrero A, Laespada-García MI, Gómez-Grande A et al (2020) Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 95(15):e2109–e2118. https://doi.org/10.1212/wnl.0000000000010282
Merello M, Bhatia KP, Obeso JA (2021) SARS-CoV-2 and the risk of Parkinson’s disease: facts and fantasy. Lancet Neurol 20(2):94–95. https://doi.org/10.1016/s1474-4422(20)30442-7
Mirsattari SM, Power C, Nath A (1998) Parkinsonism with HIV infection. Mov Disord 13(4):684–689. https://doi.org/10.1002/mds.870130413
Misra UK, Kalita J (1997) Movement disorders in Japanese encephalitis. J Neurol 244(5):299–303. https://doi.org/10.1007/s004150050090
Misra UK, Kalita J (2002) Prognosis of Japanese encephalitis patients with dystonia compared to those with parkinsonian features only. Postgrad Med J 78(918):238–241. https://doi.org/10.1136/pmj.78.918.238
Misra UK, Kalita J, Pandey S, Khanna VK, Babu GN (2005) Cerebrospinal fluid catecholamine levels in Japanese encephalitis patients with movement disorders. Neurochem Res 30(9):1075–1078. https://doi.org/10.1007/s11064-005-7414-2
Morassi M, Palmerini F, Nici S et al (2021) SARS-CoV-2-related encephalitis with prominent parkinsonism: clinical and FDG-PET correlates in two patients. J Neurol. https://doi.org/10.1007/s00415-021-10560-3
Mulder DW, Parrott M, Thaler M (1951) Sequelae of western equine encephalitis. Neurology 1(4):318–327. https://doi.org/10.1212/wnl.1.7-8.318
Murgod UA, Muthane UB, Ravi V, Radhesh S, Desai A (2001) Persistent movement disorders following Japanese encephalitis. Article. Neurology 57(12):2313–2315. https://doi.org/10.1212/WNL.57.12.2313
Navia BA, Cho ES, Petito CK, Price RW (1986) The AIDS dementia complex: II. Neuropathology. Ann Neurol 19(6):525–535. https://doi.org/10.1002/ana.410190603
Netravathi M, Pal PK, Indira DB (2012) A clinical profile of 103 patients with secondary movement disorders: correlation of etiology with phenomenology. Eur J Neurol 19(2):226–233. https://doi.org/10.1111/j.1468-1331.2011.03469.x
Ogata A, Tashiro K, Nukuzuma S, Nagashima K, Hall WW (1997) A rat model of Parkinson’s disease induced by Japanese encephalitis virus. J Neurovirol 3(2):141–147. https://doi.org/10.3109/13550289709015803
Okunoye O, Kojima G, Marston L, Walters K, Schrag A (2020) Factors associated with hospitalisation among people with Parkinson’s disease—a systematic review and meta-analysis. Parkinsonism Relat Disord 71:66–72
Ong TL, Nor KM, Yusoff Y, Sapuan S (2022) COVID-19 associated acute necrotizing encephalopathy presenting as parkinsonism and myorhythmia. J Mov Disord 15(1):89–92. https://doi.org/10.14802/jmd.21063
Pakpoor J, Noyce A, Goldacre R et al (2017) Viral hepatitis and Parkinson disease: a national record-linkage study. Neurology 88(17):1630–1633. https://doi.org/10.1212/wnl.0000000000003848
Patel H, Sander B, Nelder MP (2015) Long-term sequelae of West Nile virus-related illness: a systematic review. Lancet Infect Dis 15(8):951–959. https://doi.org/10.1016/s1473-3099(15)00134-6
Pilcher CD, Shugars DC, Fiscus SA et al (2001) HIV in body fluids during primary HIV infection: implications for pathogenesis, treatment and public health. AIDS 15(7):837–845. https://doi.org/10.1097/00002030-200105040-00004
Pilotto A, Masciocchi S, Volonghi I et al (2021) Clinical presentation and outcomes of severe acute respiratory syndrome coronavirus 2-related encephalitis: the ENCOVID Multicenter Study. J Infect Dis 223(1):28–37. https://doi.org/10.1093/infdis/jiaa609
Poser CM, Huntley CJ, Poland JD (1969) Para-encephalitic parkinsonism. Report of an acute case due to coxsackie virus type B 2 and re-examination of the etiologic concepts of postencephalitic parkinsonism. Acta Neurol Scand 45(2):199–215
Pranzatelli MR, Mott SH, Pavlakis SG, Conry JA, Tate ED (1994) Clinical spectrum of secondary parkinsonism in childhood: a reversible disorder. Pediatr Neurol 10(2):131–140. https://doi.org/10.1016/0887-8994(94)90045-0
Rao AR, Hidayathullah SM, Hegde K, Adhikari P (2022) Parkinsonism: An emerging post COVID sequelae. Idcases 27:e01388. https://doi.org/10.1016/j.idcr.2022.e01388
Rass V, Beer R, Schiefecker AJ et al (2021) Neurological outcome and quality of life 3 months after COVID-19: a prospective observational cohort study. Eur J Neurol 28(10):3348–3359. https://doi.org/10.1111/ene.14803
Raung SL, Kuo MD, Wang YM, Chen CJ (2001) Role of reactive oxygen intermediates in Japanese encephalitis virus infection in murine neuroblastoma cells. Neurosci Lett 315(1–2):9–12. https://doi.org/10.1016/s0304-3940(01)02300-x
Reid AH, Taubenberger JK, Fanning TG (2001) The 1918 Spanish influenza: integrating history and biology. Microbes Infect 3(1):81–87. https://doi.org/10.1016/s1286-4579(00)01351-4
Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R. Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathologica. 1991/06/01 1991;82(1):39–44. doi:https://doi.org/10.1007/BF00310921
Rosso ALZd, Mattos JPd, Correa RB, Nicaretta DH, Novis SAP (2009) Parkinsonism and AIDS: a clinical comparative study before and after HAART. Arq Neuropsiquiatr 67:827–830
Roy D, Song J, Awad N, Zamudio P (2021) Treatment of unexplained coma and hypokinetic-rigid syndrome in a patient with COVID-19. BMJ Case Rep. https://doi.org/10.1136/bcr-2020-239781
Sadasivan S, Sharp B, Schultz-Cherry S, Smeyne RJ (2017) Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. NPJ Parkinson’s Dis 3:18. https://doi.org/10.1038/s41531-017-0019-z
Sampson BA, Ambrosi C, Charlot A, Reiber K, Veress JF, Armbrustmacher V (2000) The pathology of human West Nile Virus infection. Hum Pathol 31(5):527–531. https://doi.org/10.1053/hp.2000.8047
Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS (2007) Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci USA 104(43):17140–17145. https://doi.org/10.1073/pnas.0705837104
Santerre M, Arjona SP, Allen CN, Callen S, Buch S, Sawaya BE (2021) HIV-1 Vpr protein impairs lysosome clearance causing SNCA/alpha-synuclein accumulation in neurons. Autophagy 17(7):1768–1782. https://doi.org/10.1080/15548627.2021.1915641 (PMID-33890542)
Sardar AM, Czudek C, Reynolds GP (1996) Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. NeuroReport 7(4):910–912. https://doi.org/10.1097/00001756-199603220-00015
Schacker T, Collier AC, Hughes J, Shea T, Corey L (1996) Annals of internal medicine: clinical and epidemiologic features of primary HIV infection. Ann Intern Med 125(4):257–264. https://doi.org/10.7326/0003-4819-125-4-199608150-00001
Scherbaum R, Kwon EH, Richter D et al (2021) Clinical profiles and mortality of COVID-19 inpatients with Parkinson’s disease in Germany. Mov Disord 36(5):1049–1057
Sejvar JJ (2016) West Nile virus infection. Microbiol Spectr 4(3):10. https://doi.org/10.1128/microbiolspec.EI10-0021-2016
Sejvar JJ (2014) Clinical manifestations and outcomes of West Nile virus infection. Viruses. 6(2):606–623. https://doi.org/10.3390/v6020606
Sivan M, Taylor S (2020) NICE guideline on long covid. British Medical Journal Publishing Group
Smeyne RJ, Noyce AJ, Byrne M, Savica R, Marras C (2020) Infection and risk of parkinson’s disease. J Park Dis 11(1):31–43. https://doi.org/10.3233/jpd-202279 (PMID-33361610)
Solomon T, Fisher AF, Beasley DWC et al (2003) Natural and nosocomial infection in a patient with west nile encephalitis and extrapyramidal movement disorders. Clin Infect Dis 36(11):e140–e145. https://doi.org/10.1086/374936
Su TH, Yang HC, Tseng TC et al (2019) Antiviral therapy in patients with chronic hepatitis C is associated with a reduced risk of parkinsonism. Mov Disord 34(12):1882–1890. https://doi.org/10.1002/mds.27848 (PMID-31505068)
Sulzer D, Antonini A, Leta V et al (2020) COVID-19 and possible links with Parkinson’s disease and parkinsonism: from bench to bedside. NPJ Parkinson’s Dis 6:18. https://doi.org/10.1038/s41531-020-00123-0
Takahashi M, Yamada T, Nakajima S, Nakajima K, Yamamoto T, Okada H (1995) The substantia nigra is a major target for neurovirulent influenza A virus. J Exp Med 181(6):2161–2169. https://doi.org/10.1084/jem.181.6.2161
Tanaka H, Park C-H, Ninomiya A et al (2003) Neurotropism of the 1997 Hong Kong H5N1 influenza virus in mice. Vet Microbiol 95(1–2):1–13. https://doi.org/10.1016/s0378-1135(03)00132-9
Taubenberger JK (2006) The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc 150(1):86–112
Tiraboschi P, Xhani R, Zerbi SM et al (2021) Postinfectious neurologic complications in COVID-19: a complex case report. J Nucl Med 62(8):1171–1176. https://doi.org/10.2967/jnumed.120.256099
Toovey S, Jick SS, Meier CR (2011) Parkinson’s disease or Parkinson symptoms following seasonal influenza. Influenza Other Resp 5(5):328–333. https://doi.org/10.1111/j.1750-2659.2011.00232.x (PMID-21668692)
Tsai H-H, Liou H-H, Muo C-H, Lee C-Z, Yen R-F, Kao C-H (2016) Hepatitis C virus infection as a risk factor for Parkinson disease. Neurology 86(9):840–846. https://doi.org/10.1212/wnl.0000000000002307 (PMID-26701382)
Tse W, Cersosimo MG, Gracies JM, Morgello S, Olanow CW, Koller W (2004) Movement disorders and AIDS: a review. Parkinsonism Relat Disord 10(6):323–334. https://doi.org/10.1016/j.parkreldis.2004.03.001
Turtle L, Solomon T (2018) Japanese encephalitis — the prospects for new treatments. Nat Rev Neurol 14(5):298–313. https://doi.org/10.1038/nrneurol.2018.30
Tysnes OB, Storstein A (2017) Epidemiology of Parkinson’s disease. J Neural Transm (vienna, Austria : 1996) 124(8):901–905. https://doi.org/10.1007/s00702-017-1686-y
van Gent M, Braem SG, de Jong A et al (2014) Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog 10(2):e1003960. https://doi.org/10.1371/journal.ppat.1003960
van Wamelen DJ, Leta V, Johnson J et al (2020) Drooling in Parkinson’s disease: prevalence and progression from the non-motor international longitudinal study. Dysphagia 35(6):955–961
Vilensky JA, Gilman S, McCall S (2010a) A historical analysis of the relationship between encephalitis lethargica and postencephalitic parkinsonism: a complex rather than a direct relationship. Mov Disord 25(9):1116–1123. https://doi.org/10.1002/mds.22908
Vilensky JA, Gilman S, McCall S (2010b) Does the historical literature on encephalitis lethargica support a simple (direct) relationship with postencephalitic Parkinsonism? Mov Disord 25(9):1124–1130. https://doi.org/10.1002/mds.22991
Vincent FM, Myers WG (1978) Poliomyelitis and Parkinsonism. N Engl J Med 298(12):688–689. https://doi.org/10.1056/NEJM197803232981216
Walters JH (1960) Postencephalitic parkinson syndrome after meningoencephalitis due to coxsackie virus group B, type 2. N Engl J Med 263(15):744–747. https://doi.org/10.1056/nejm196010132631507
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA (2004) Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10(12):1366–1373. https://doi.org/10.1038/nm1140
Wang H, Liu X, Tan C et al (2020) Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case-control studies. Brain Behav 10(3):e01549. https://doi.org/10.1002/brb3.1549 (PMID-32017453)
Weissenborn K, Ennen JC, Bokemeyer M et al (2006) Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut 55(11):1624–1630. https://doi.org/10.1136/gut.2005.080267
Werring DJ, Chaudhuri KR (1996) Human immunodeficiency virus-related progressive multifocal leukoencephalopathy presenting with an akinetic rigid syndrome. Mov Disord 11(6):758–761. https://doi.org/10.1002/mds.870110633
Wheeler EDA, Achim CL, Ayyavoo V (2006) Immunodetection of human immunodeficiency virus type 1 (HIV-1) Vpr in brain tissue of HIV-1 encephalitic patients. J Neurovirol 12(3):200–210. https://doi.org/10.1080/13550280600827377
Wijarnpreecha K, Chesdachai S, Jaruvongvanich V, Ungprasert P (2018) Hepatitis C virus infection and risk of Parkinson’s disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol 30(1):9–13. https://doi.org/10.1097/MEG.0000000000000991
Worldmeter.info (2021) COVID-19 CORONAVIRUS PANDEMIC. Updated 24/10/2021. https://www.worldometers.info/coronavirus/. Accessed 14 Feb 2022
Woulfe J, Hoogendoorn H, Tarnopolsky M, Muñoz DG (2000) Monoclonal antibodies against Epstein-Barr virus cross-react with α-synuclein in human brain. Article. Neurology 55(9):1398–1401. https://doi.org/10.1212/WNL.55.9.1398
Wu WYY, Kang KH, Chen SLS et al (2015) Hepatitis C virus infection: a risk factor for Parkinson’s disease. J Viral Hepatitis 22(10):784–791. https://doi.org/10.1111/jvh.12392 (PMID-25608223)
Xing F, Marsili L, Truong DD (2022) Parkinsonism in viral, paraneoplastic, and autoimmune diseases. J Neurol Sci 433:120014. https://doi.org/10.1016/j.jns.2021.120014
Zou S, Foster GA, Dodd RY, Petersen LR, Stramer SL (2010) West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis 202(9):1354–1361. https://doi.org/10.1086/656602
Acknowledgements
The views expressed are those of the authors and not necessarily those of the NHS, NIHR or Department of Health. The authors acknowledge the support of the International Parkinson and Movement Disorder Society Non-Motor PD Study Group, the NIHR London South Clinical Research Network and the NIHR Biomedical Research Centre. This article represents independent collaborative research part funded by the NIHR Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leta, V., Urso, D., Batzu, L. et al. Viruses, parkinsonism and Parkinson’s disease: the past, present and future. J Neural Transm 129, 1119–1132 (2022). https://doi.org/10.1007/s00702-022-02536-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-022-02536-y